 علوم القطيف مقالات علمية في شتى المجالات العلمية
علوم القطيف مقالات علمية في شتى المجالات العلمية
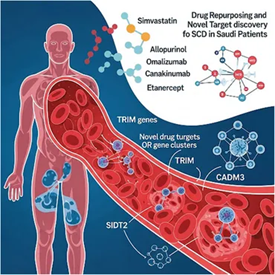
Genomics-driven drug repurposing and novel targets identification for sickle cell disease in Saudi patients
(دراسة رائدة مستندة إلى علم الجينوم لإعادة توظيف أدوية مستخدمة لعلاج أمراض أخرى، لعلاج مرضى فقر الدم المنجلي السعوديين)
[المؤلفون؛ على الغبيشي، محمد الشبيب، ويجيسينجها ديانجان، محمد العوض، سعاد الشمري، خليفة الراجح، منى الخيري، عماد الإسلام، وأحمد العسكر]
(الناشر؛ مجلة فرونتيرز في المعلوماتية الحيوية)
{راجع الترجمة وقدم للدراسة الدكتور محمد آل شبيب، استشاري علم الجينات الدوائية، عالم أبحاث، مركز الملك عبد الله العالمي للأبحاث الطبية، الرياض}
مقدمة الدكتور محمد آل شبيب
تم بحمد الله مؤخرًا إكمال ونشر بحثنا حول تحديد تطبيقات جديدة لأدوية متوفرة حاليًا في الأسواق للمساعدة في علاج داء الخلايا المنجلية (السكلسل)، وذلك بناءً على معرفة متغيرات جينية جزيئية اكتشفناها مؤخرًا.
الدراسة المترجمة أدناه هي دراسة نوعية تُظهر كيفية إعادة توظيف بعض الأدوية المعتمدة والمعروفة باستخدامها في علاج أمراض أخرى، للاستفادة منها لاستهداف مسارات جينية محددة ومؤثرة في مرض الخلايا المنجلية. ركز البحث على عدة مؤشرات جزيئية أساسية ارتبطت بشدة المرض، بما في ذلك جينات متعددة تؤثر في بعض المسارات المساهمة في الالتهابات المزمنة لدى مرضى السكلسل وانغلاق الأوعية الدموية.
الأبعاد السريرية: يمثل هذا البحث تقدمًا مهمًا في خيارات علاج المرض، ويوفر إمكانيات تشمل:
– إيجاد بدائل علاجية أكثر سهولة مقارنة بالعلاجات الجينية الحديثة مثل (Casgevy) و (Lyfgenia) الباهظة الثمن
– تقليل تكاليف العلاج، وذلك بالاستفادة من أدوية معتمدة رخيصة ومصادق عليها سابقًا
– سرعة التطبيق العملي نظرًا لسجلات السلامة المعروفة لهذه الأدوية
– توفير حلول علاجية موجهة بحسب الفحص الجيني لكل مريض.
يرتكز هذا البحث على تطورات حديثة في فهم المؤشرات الجينية للمرض، بما يشمل دراسات الارتباط على مستوى الجينوم التي أجراها فريقنا البحثي في دراسة سابقة.
أضاف المترجم تعريفات ونصوص وبعض الشروح المناسبة بين مزدوجين، للتعريف ببعض المصطلحات الطبية مع إدراج بعض الأمثلة لزيادة الإيضاح.
( الورقة المترجمة )
خلفية الدراسة:
داء الخلايا المنجلية أو فقر الدم المنجلي (سكلسل SCD)، والذي يعرف أيضًا بداء الأنيميا المنجلية، هو اضطراب دم وراثي يتميز بانحلال الدم المزمن (انحلال أنيمي بسبب تحلل كريات الدم الحمراء)، والالتهاب، ونوبات انسداد الأوعية الدموية (VOC) بسبب التصاق وتكتل الخلايا المنجلية داخل الأوعية الدقيقة، مما يؤدي إلى معاناة المصاب بحالات مرضية ثانوية وانخفاض في متوسط عمره المتوقع. تتوفر حاليًا خيارات علاجية فعالة، لكنها محدودة؛ بيد أن نتائج فحوص جينية حديثة (مصممة للتعرف على تغيرات في الحامض النووي -الدنا- للفرد) من عينة منحصرة في مرضى فقر الدم المنجلي السعوديين (غير ممثلة على الصعيد العالمي) تبعث بأمل جديد للتنبؤ بعلاجات موجهة جزيئيًا (علاجات مصممة لاستهداف التغيرات الجزيئية في جينات معينة) في مرضى فقر الدم المنجلي. هدفت هذه الدراسة إلى التعرف على أدوية معتمدة سابقًا لعلاج أمراض أخرى، والتي يعتقد أنها مناسبة لعلاج هذا المرض الوراثي بناءً على تفاعلاتها مع المتغيرات الجينية المرتبطة بمرض فقر الدم المنجلي، واكتشاف جينات جديدة لها مواضع يمكن أن يرتبط بها الدواء ويغير وظيفتها ويجعلها قابلة للتدخل العلاجي ضمن المسارات أو الشبكات الجينية التي تجعل المرض أكثر حدة أو أكثر سوءً (ما يعرف بـ druggability)، وذلك باستخدام بيانات دراسة الترابط الجينومي الكامل (GWAS) المعمولة على مرضى فقر الدم المنجلي السعوديين.
مقدمة
مرض فقر الدم المنجلي (SCD) هو مجموعة من اضطرابات الدم الوراثية الحادة، يُسببه وجود متغير (طفرة) قي الهيموغلوبين (rs334 c.20 A>T) في جين (HBB) (أي متغير الحمض النووي المسبب لفقر الدم المنجلي رقم 334 في الموضع رقم 20 في ذلك الجين، بحيث يتغير الحمض الأميني الأدينين إلى الثايمين)، مما يُسبب نشوء هيموغلوبين غير طبيعي يسمى هيموغلوبين S والذي ينتج تشوهًا “تمنجلًا” لكريات الدم الحمراء(1).
يؤدي هذا التمنجل إلى سلسلة مضاعفات وعائية لاحقة، بما فيها نوبات انسداد الأوعية الدموية، وتخثر للدم، واحتشاءات (نخر) في أعضاء متعددة، وذلك بموت الأنسجة فيها بسبب نقص التروية، مما يُسهم في شدة مرض فقر الدم المنجلي سريريًا (والذي يؤثر سلبا في حياة المصاب) من مرحلة الطفولة المبكرة فصاعدًا(1، 2). وتتجلى خطورة هذه المضاعفات بشكل أكثر في انخفاض متوسط العمر المتوقع لدى المصابين بفقر الدم المنجلي مقارنةً بغير المصابين في عموم السكان(3).
من منظور الصحة العامة، يُشكل مرض فقر الدم المنجلي (SCD) عبئًا عالميًا كبيرًا. فبينما تفيد بعض التقديرات المنشورة إلى وجود أكثر من 7 ملايين شخص مُصاب بالمرض حول العالم(4، 5)، تُقدّر منظمة الصحة العالمية (WHO) أن حوالي 300 ألف رضيع يُولدون سنويًا مصابين بهذه الحالة. ويُقدّر أن نيجيريا وحدها تُسجّل (نصف هذا العدد) حوالي 150 ألف مولود مصاب بمرض فقر الدم المنجلي سنويًا، ما يجعلها الدولة الأعلى عالميًا في معدل الإصابة بفقر الدم المنجلي(6).
ولكن، نظرًا لارتفاع معدلات الوفيات في مرحلة الطفولة في الكثير من المناطق المُوبوءة والفشل في تشخيص المرض أو تشخيصه بشكل خاطئ، لا سيما في جنوب الصحراء الكبرى الأفريقية وجنوب آسيا، فإن العدد الحقيقي للمُصابين بفقر الدم المنجلي ما يزال غير مؤكد ومُثارًا للجدل بين الباحثين في الأدبيات العلمية المنشورة.
كما تعاني عدد من الدول، بما فيها دول شبه القارة الهندية ودول الأمريكتين ودول منطقة البحر الأبيض المتوسط ودول الشرق الأوسط من الزيادة المطّردة لهذا المرض، بالرغم من أن البيانات الوبائية الدقيقة غالبًا ما تكون محدودة بسبب عدم كفاية الرصد والمراقبة وارتفاع معدلات وفيات الأطفال من الناحية التاريخية، بسبب نقص العناية الطبية(7).
انعكاسًا لعبء هذا المرض عالميًا، تواجه المملكة العربية السعودية مستوى عبء مرتفع بشكل ملحوظ من جراء الإصابة بمرض فقر الدم المنجلي، حيث تصل معدلات حاملي نسخة واحدة من الجين المسبب للمرض (HbSA) الموثقة إلى 27% (وإن لم تظهر عليهم بالضرورة أعراض المرض وقد يعيشون طبيعيًا وبصحة جيدة)، بالرغم من أن دراسات قد أشارت إلى تقديرات عن نسبة انتشار المرض بلغت حوالي 2.6% (هؤلاء هم مرضى فقر الدم المصابون به وراثيًا ويحملون نسختين من الجين المسبب للمرض (HbSS) وتظهر عليهم أعراض المرض)(7، 8).
نسبة الانتشار هذه لا تُمثل متوسط الانتشار في عموم مناطق السعودية، بل قد تختلف اختلافًا كبيرًا باختلاف المناطق، مع ملاحظة ارتفاع المعدلات في المحافظات الشرقية والجنوبية الغربية، بحيث يعد مرض فقر الدم المنجلي فيها أكثر انتشارًا من غيرها من المحافظات الأخرى. ومن المرجح أن يتفاقم هذا العبء الكبير بسبب ارتفاع معدلات زواج الأقارب (حيث تصل إلى معدلات تزيد على 50%)، والتي تتركز فيها المتغيرات الوراثية للمرض(8، 10). في بعض المناطق بالمملكة، ما يقرب من 90% من حاملي المرض أصروا على الإقدام على الزواج من نظرائهم، بالرغم من وجود توصيات برنامج فحص ما قبل الزواج بعدم الإقدام(8).
الأدوية المعتمدة لعلاج مرض فقر الدم المنجلي ما زالت غير متوفرة بشكل كافٍ، ولا تلبي حاجة العدد المتزايد من المرضى. في الأصل، دواء هيدروكسي يوريا (hydroxyurea) كان هو العلاج الوحيد المعتمد من قِبل إدارة الغذاء والدواء الأمريكية (FDA) للراشدين (18 سنة وأكبر) والأطفال المصابين بمرض فقر الدم المنجلي. منذ عام 2017، تطور المشهد العلاجي وتغير الوضع العام للعلاجات بعد أن وافقت إدارة الغذاء والدواء الأمريكية على اعتماد ثلاثة أدوية جديدة تستهدف جوانب مختلفة من فسيولوجيا مرض فقر الدم المنجلي، بما فيها دواء غلوتامين (L-glutamine)، وكريزانليزوماب (crizanlizumab)، وفوكسيلوتور(11، 12) (voxelotor)، ولكن التطورات الأخيرة أبرزت بعض الصعوبات في استخدام هذه الأدوية.
والجدير بالذكر أنه اعتبارًا من سبتمبر، 2024، سُحب فوكسيلوتور طواعيةً من الأسواق العالمية بسبب مخاوف تتعلق بسلامة الدواء، وفي مايو 2023، تم سحب كريزانليزوماب من السوق الأوروبية بسبب ضعف مفعوليته(13، 14). وبالرغم من سحب الوكالة الأوروبية للأدوية (EMA) موافقتها لهذا الدواء بعد مراجعة بيانات تجربة (STAND) الجديدة السلبية التي لم تُثبت أي فائدة مضافة، استمرت إدارة الغذاء والدواء الأمريكية (FDA) في موافقتها على اعتماد كريزانليزوماب لعلاج فقر الدم المنجلي، لكنها ما زالت تنتظر مراجعة شاملة للبيانات الجديدة والتي تبيّٓن أنها غير حاسمة بعد.
تُبرز هذه الأحداث تعقيدات علاج مرض فقر الدم المنجلي، وتُؤكد على الأهمية الحاسمة لمواصلة الدراسات بهذا الخصوص، والمراقبة الدقيقة لسلامة أي علاجات مستقبلية في الأمد الطويل، واستمرار مراجعة التقييم الدوري للعلاجات المعتمدة بالفعل.
بالرغم من أن زراعة النخاع العظمي (BMT) قد يكون علاجًا ممكنًا لمرض فقر الدم المنجلي، إلا أن هناك تحديات معتبرة تقف حجر عثرة أمام استخدام هذه التقنية للعلاج، بما فيها ارتفاع تكلفتها المالية، وعدم توفر المتبرعين المناسبين لنقل / زرع النخاخ، والمخاطر المصاحبة لها. الموافقة الأخيرة على اعتماد أول علاجين خلويين وجينيين (يعملان على تغيير الخلايا أو الجينات)، وهما (Casgevy) و(Lyfgenia) ، في 8 ديسمبر 2023، أحدث تقدمًًا كبيرًا في علاج فقر الدم المنجلي(15).
المملكة العربية السعودية هي أول دولة في العالم استخدمت علاج (Casgevy) بعد اعتماده ونجحت في معالجة خمسة من المرضى حتى الآن. ومع ذلك، ما تزال هذه العلاجات المتقدمة غير متاحة للكثير من مرضى فقر الدم المنجلي، نظرًا لتكاليفها المالية الباهظة جدًاً حيث تبلغ تكلفة (Casgevy) مليونين ومائتي ألف دولار أمريكي للمريض الواحد، بينما تبلغ تكلفة (Lyfgenia) ثلاثة مليون ومائة ألف دولار أمريكي للمريض الواحد. تُعد هذه العلاجات الجينية من أغلى الأدوية المُعتمدة على الإطلاق، وتمثل تحديًا اقتصاديًا كبيرًا للمرضى والأنظمة الصحية على حد سواء(16).
يُبرز هذا الوضع الحاجة المُلحة لإجراء المزيد من الدراسات تتناول اكتشاف أدوية مبتكرة تُوفر خيارات علاجية أكثر أمانًا ومفعولية وبأسعار معقولة لمرضى فقر الدم المنجلي.
علاوةً على ذلك، تُمثل الطبيعة الجينية المُعقدة لمرض فقر الدم المنجلي، إلى جانب المتغيرات الجينية المُرتبطة بالمرض والتي تم التعرف عليها مؤخرًا من خلال دراسة الترابط الجينومي الكامل (GWAS)، فرصةً مثاليةً لتوجيه الجهود تجاه استكشاف امكانية استخدام الأدوية المعتمدة سابقًا لعلاج أمراض أخرى وتطوير علاجات جديدة لفقر الدم المنجلي(18، 19).
دراسات الترابط الجينومي الكامل الأخيرة على مجموعة من مرضى فقر الدم المنجلي السعوديين – لا سيما تلك التي أجراها الدكتور الشبيب وآخرون(20) والدكتور الغبيشي وآخرون(19) – ساهمت في توضيح المشهد الجيني، مُثبتةً أن المتغيرات التي لاحظناها في المرضى السعوديين خاصةً هي جزئيًا أكثر شيوعًا بين السكان السعوديين، وذلك لوجود علامات فريدة ومشتركة بينهم مقارنة بسكان من أصول أفريقية.
هذه النتائج تفيد بأن اختلافات جينومية ملحوظة ناجمة عن النمط المميز “العربي-الهندي” السائد في شرق المملكة العربية السعودية، والذي يختلف في انتشاره وتبعاته السريرية عن الأنماط الخاصة الأفريقية (بنين، السنغال، الكاميرون، ومجموعات البانتو العرقية في جنوب أفريقيا)، حيث أن النمط المميز يعني توليفة من الطفرات في مجموعة جينات مرتبطة وراثيًا وتحتل مواضع متجاورة على الكرموسوم.
إعادة توظيف الأدوية المستخدمة لعلاج أمراض أخرى تُمثل منهجيةً لاقتراح استخدامات جديدة للأدوية المعتمدة والمتوفرة في الأسواق حاليًا، استنادًا إلى تفاعلاتها مع الجينات الرئيسة التي تُعدّل المسارات الخلوية (أي التي تؤثّر في طريقة تواصل الخلايا مع بعضها مما يحدث سلسلة من التفاعلات داخل الخلية تتحكم في وظائف الخلية، مثل نموها وتخصصها وموتها)، وقد تهدف إلى تحسين أعراض المرض، وذلك بخفض حدة الأعراض، وتأخير تقدم المرض، والحد من مضاعفاته، والبقاء على قيد الحياة(20). وقد استُخدمت هذه المقاربة الحوسبية، التي تعتمد على تحليلات الجينات المستهدفة، والتي تستخدم أساليب حوسبية لتحليل البيانات الجينية لاستكشاف امكانية إعادة توظيف أدوية عديدة تستخدم حاليًا لعلاج حالات مرضية مُختلفة(21).
دراستنا ترتكز على إعادة توظيف الأدوية المستخدمة حاليا واكتشاف مركبات جديدة لمرض فقر الدم المنجلي، والتي ستخضع بعد ذلك للتجارب السريرية. يمكن لهذه التطورات أن تُحدث ثورة في إدارة مرض فقر الدم المنجلي، وتُعزز المعرفة العلمية والقدرات التقنية والممارسات السريرية. من خلال استكشاف أهداف جينية مؤثرة في المرض يمكن تصميم أدوية جديدة مناسبة للتفاعل معها. من شأن هذه الجهود أن تُفضي إلى التوصل واكتشاف علاجات رائدة، وتُرسي معايير جديدة في هذا المجال، وتُعزز الابتكار في الطب الشخصي (العلاج الموجه لشخص بعينه).
بالإضافة إلى تمنجل خلايا الدم الحمراء بسبب متغير (HbS) ، يُظهر طيف من أعراض مرض فقر الدم المنجلي تباينًا سريريًا واسعًا من ناحية شدة الأعراض وتفاقمها متأثرةً بوجود متغيرات جينية متعددة(22). كشفت دراسات جينية سابقة على مرضى الأنيميا المنجلية عن تعدد أشكال الطفرات التي تُنظم مسارات جزيئية مختلفة للمرض، بما في ذلك انحلال الدم، والالتهاب، والإجهاد التأكسدي، واختلال وظائف الأوعية الدموية(22، 23).
تشمل الأمثلة المتغيرات المنظمة للهيموغلوبين الجنيني (HbF) في (BCL11A) (الجين اﻟذي ﯾﺗﺣﮐم ﻓﻲ إﻧﺗﺎج اﻟﮭﯾﻣوﺟﻟوﺑﯾن اﻟﺟﻧﯾﻧﻲ ﻓﻲ اﻟﺟﺳم) و(HBS1L-MYB) (منطقة جينية تنظيمية تقع بين جين (MYB) وبين جين (HBS1L) تؤثر في صفات الدم وتؤثر في جين (MYB)، الذي له دور في انتاج خلايا الدم الحمراء وصفائح الدم. التغيرات في هذه المنطقة الجنينية لها علاقة بشدة مرض فقر الدم المنجلي)، بالإضافة إلى تعدد أشكال السايتوكينات المسببة للالتهابات في محفز نخر الورم (سايتوكاين) “TNF-α” (المسبب للالتهاب) وجزيئات الالتصاق الخلوية (VCAM-1)، والتي ترتبط بزيادة احتمال الإصابة بالسكتة الدماغية(24).
هذه الأفكار الجينية لا تزودنا بأطر ضرورية فحسب لإعادة استخدام الأدوية الحالية لاستهداف آليات الخلايا المنجلية الناشئة (مثل التصاق الخلايا ببطانة الأوعية الدموية وانحلال الخلايا الذي يتسبب في التحفيز الضار للجهاز المناعي)، بل تمهد الطريق أيضًا لتطوير علاجات مبتكرة ومصممة خصيصًا يمكنها تحسين مخرجات المرضى (الهناء النفسي والصحة العقلية وجودة الحياة) بشكل كبير(25).
نقص العلاجات الفعالة لمرض فقر الدم المنجلي شجع على استكشاف استراتيجيات علاجية بديلة، مثل إعادة توظيف الأدوية القائمة على المسارات والشبكات البيولوجية (لمعرفة المسارات والشبكات البيولوجية لاكتشاف أدوية لعلاج أمراض جديدة)، أو النمط الظاهري (المقتصر على التغيرات الظاهرية في الخلية القابلة للملاحظة لمعرفة تأثير الدواء)، أو التحليل الحاسوبي (استخدام الأساليب الحاسوبية وعلم المعلومات البيولوجية للتنبؤ بأدوية جديدة)، والاستفادة من مكتبات المركبات الكيميائية الحالية أو الأدوية المعتمدة لاستهداف آليات مرض فقر الدم المنجلي المستجدة(26، 27، 28).
دمج البيانات الجينية من الدراسات الجينومية عالية الإنتاجية الحديثة في مجموعات متنوعة من مرضى فقر الدم المنجلي يجعلنا متمكنين من اكتشاف أهداف (جينات) جديدة لها مواضع يمكن أن يرتبط بها الدواء ويغير وظيفتها ويجعلها قابلة للتدخل العلاجي، ويعطي أولوية للأدوية المرشحة للعلاج للفحص السريريّ(20، 22) (ما يصطلح عليه بـ (druggability) وهي قابلية البروتينات الناتجة من جينات محددة من الارتباط والتفاعل مع أدوية معينة).
معظم الدراسات السابقة ركزت بشكل رئيس على الأصول الأفريقية أو الأمريكية الأفريقية، بالإضافة إلى دراسة محدودة للفئات السكانية الأخرى المعرضة للإصابة، مثل تلك الموجودة في منطقتي البحر الأبيض المتوسط والشرق الأوسط(29).
مؤخرًا، كشفت مجموعتنا البحثية عن متغيرات جينية في عينات من سعوديين تُسبب مضاعفات الانصمام الخثاري (انفصال جلطة أو خثرة دم من موقعها الأصلي وتحركها في مجرى الدم وعرقلته أو سده) لدى مرضى فقر الدم المنجلي، على النقيض من علامات حيوية حُددت في مجموعات أخرى(20). إضافةً إلى ذلك، حددت نتائج بياناتنا الحالية عدة مواضع في جينات معروفة مرتبطة بمسارات مرضية جزيئية قد تؤثر في شدة مرض فقر الدم المنجلي، مما يُؤكد الحاجة إلى تدخلات مُصممة خصيصًا لهؤلاء المرضى(19).
توسيع نطاق توصيف الأسس الجزيئية الخاصة بكل فئة سكانية من خلال علم الجينوم الشامل له ميزتان رئيسيتان: فهو يكشف عن أهداف علاجية جديدة خاصة بالسعوديين، ويُوضح التباين في الآليات المُمرضة (أي التنوع في كيفية نشوء مسببات الأعراض المختلفة للمرض) عبر طيف مرضى فقر الدم المنجلي عالميًا (أي انتشاره عالميًا والخصائص المميزة للمصابين بفقر الدم المنجلي).
لذلك، هدفنا في هذه الدراسة إلى الاستفادة من نتائج تحليلنا للترابط الجينومي الكامل، بالتزامن مع اكتشاف أهداف (جينات) جديدة لها مواضع يمكن أن يرتبط بها الدواء ويغير وظيفتها ويجعلها قابلة للتدخل العلاجي (ما يصطلح عليه بـ druggability)، مع التركيز تحديدًا على مرضى خلايا الدم المنجلية (SCD) السعوديين، لاقتراح خيارات علاجية جديدة.
هدفنا الواسع والبعيد الأمد هو وضع خارطة طريق مدروسة وموجهة جينيًا تُسرّع من تطوير علاجات مبتكرة ومصممة خصيصًا لهذه الفئة من مرضى فقر الدم المنجلي التي لم تلقْ من العناية ما تستحق في السابق. كما نحاول أن نستخدم هذه المقاربة الشمولية القائمة على المعرفة الجزيئية إلى تحسين وتسريع جهود اكتشاف الأدوية المستقبلية.
2 منهجية العمل البحثي
2.1 خط عملية تطوير إعادة استخدام الأدوية المعتمدة وتطوير أدوية جديدة
استخدمنا بيانات مستمدة من تحليلنا السابق لـ دراسة الترابط الجينومي الكامل لتحليل المتغيرات الجينية المرتبطة بمرض فقر الدم المنجلي في عينات من سكان المملكة العربية السعودية(19). ركز تحليلنا على تحديد التفاعلات الممكنة بين الجينات والأدوية للتعرف على الأدوية المرشحة التي يمكن إعادة استخدامها لعلاج فقر الدم المنجلي، واقتراح مركبات جديدة لتطويرها كخيارات علاجية مستقبلية. التحليل في هذه الدراسة جرى وفقًا للمنهجية العملية المستخدمة في دراسة نشرت سابقًا (الشكل 1)(30).
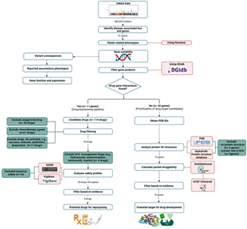
2.2 فحص الجينات المرتبطة بشدة مرض فقر الدم المنجلي
ركزنا على 31 من الجينات المرتبطة بشدة مرض فقر الدم المنجلي والتي نشرها الغبيشي وآخرون(19). خضعت هذه الجينات للاختبار بناءً على دورها في المسارات المرضية الرئيسة لمرض خلايا الدم المنجلية، بما فيها السيطرة على الالتهاب (والمحافظة عليها عند مستوى معين وإيقافها في النهاية)، ووظيفة الأوعية الدموية (مدى جودة الأوعية الدموية في توزيع الدم إلى سائر أنحاء الجسم)، وتحسين الخلايا البطانية (الطريقة التي تحسن بها الطبقة الرقيقة داخل الأوعية الدموية وظيفة الأوعية الدموية)، وغيرها من العمليات البيولوجية المرتبطة بالمرض.
تضمنت معايير الاختيار تحليل مسارات الجينات المحددة (وهي طريقة حاسوبية للبحث عن مسارات بيولوجية لقائمة جينات لفهم وظائفها البيولوجية) ومراجعة شاملة للأدبيات المنشورة في هذا الخصوص، ما أثبت العلاقة الوظيفية لهذه الجينات بالأنماط السريرية لمرض فقر الدم المنجلي.
2.3 تحليل تفاعل الدواء مع الجينات لإعادة استخدام الأدوية المعتمدة سابقًا
تم اختيار المنتجات الجينية بناءً على قدرتها على التفاعل مع الأدوية الطبية باستخدام قاعدة بيانات تفاعل الدواء مع الجينات (DGIdb) الإصدار رقم 5.0، التي تجمع البيانات من مصادر متعددة، بما فيها (DrugBank) وقاعدة علم الصيدلة الجيني المعرفية (PharmGKB)، لاكتشاف فرص إعادة استخدام الأدوية التي يمكن أن تكون مفيدة لعلاج فقر الدم المنجلي(31).
قاعدة البيانات تنطوي على شرح تفصيلي لتفاعلات الدواء مع الجينات، والجينات التي لها مواضع (جيوب Pockets) يمكن أن يرتبط بها الدواء ويغير وظيفتها ويجعلها قابلة للتدخل العلاجي (gene druggability)، بما في ذلك الأدوية المعتمدة سابقًا والمركبات التجريبية [مادة خضعت للاختبار في المختبر ووافقت عليها إدارة الغذاء والدواء الأمريكية (FDA) لتجربتها سريريًا على البشر]. ونظرًا لأن المنتجات الجينية (البروتينات) يمكن أن تتفاعل مع أدوية متعددة وخلصنا لقائمة طويلة من الأدوية المقترحة، فقد استبعدنا أولًا الأدوية غير المتاحة سريريًا (مثل الأدوية التي تم إيقاف استخدامها أو المواد قيد الدراسة في التجارب السريرية لمعرفة ما إذا هي آمنة وفعالة للعلاج) نظرًا لعدم قابليتها للتطبيق السريري المباشر (أي عدم إمكانية استخدام النتائج في التطبيقات السريرية الحالية مباشرة دون تأخير). ثم ربطت الأدوية المعتمدة بالجينات ذات الأهمية وتأثيرها المعروف في مسارات المرض، مستبعدين المواد غير الدوائية.
خلال عملية التصفية الأولية، أعطينا الأولوية للأدوية التي تستهدف مضاعفات فقر الدم المنجلي الشائعة (من أمثال الالتهابات، واختلال وظائف الأوعية الدموية، والاضطرابات المناعية)، لأن هذه المسارات تؤثر بشكل مباشر في شدة فقر الدم المنجلي. كما استُبعدت المواد المستخدمة في التشخيص، والفيتامينات، والمكملات الغذائية، والأدوية التي استبدلت (ببدائل أكثر فعالية).
من بين قائمة الأدوية التي جرت تصفيتها في المرحلة السابقة أّعطينا الأولية ثانية وبشكل أكثر لبعض الأدوية بناءً على مدى أمانها المتوقع (الأقل من حيث أثارها الجانبية)، باستخدام برنامج (VigiBase) وهو أكبر سجل عالمي لمعلومات الآثار الجانبية للأدوية الذي يشرف عليه مركز أوبسالا السويدي لصالح منظمة الصحة العالمية(32)، وقاعدة بيانات موارد آثار الأدوية الجانبية (SIDER 4.1). استبعدنا الأدوية التي لها آثار جانبية شديدة مثل العلاجات الكيميائية والأدوية التي تتميز بمستوى سمية خطير بناءً على آثارها الضارة جدًا بالسلامة.
كما استُبعدت العلاجات التي تُشكل إصابة محتملة بفقر الدم الانحلالي، باعتباره اجراءً وقائيًا لمنع تفاقم انحلال الدم لدى مرضى فقر الدم المنجلي. للتعرف على قائمة الأدوية المحتملة بشكل شمولي، استخدمنا أداة البحث عن تفاعلات المواد الكيميائية (STITCH) وقواعد بيانات منظمة الصحة العالمية (WHO) لتوحيد تسمية الأدوية لتتوافق مع الأسماء الدولية غير مسجلة المُلكية (INN) والأسماء المعتمدة في الولايات المتحدة (USAN)، ولتعيين رموز المواد الكيميائية العلاجية التشريحيةACT (34، 35) أعطينا الأولوية لتجنب الأدوية التي قد تزيد من شدة مرض فقر الدم المنجلي، وخاصةً تلك التي تنطوي على احتمال الاصابة بالعدوى، أو التدهور الإدراكي، أو عدم استقرار ضغط الدم، نظرًا لارتفاع احتمالية تعرض مرضى فقر الدم المنجلي لهذه المضاعفات.
3. النتائج
3.1 أدوية مرشحة واعدة يمكن إعادة استخدامها لعلاج فقر الدم المنجلي
من بين 31 جينًا مقترحًا باعتبارها مُعدِّلات أو عوامل تعمل على تغيير حدة أعراض مرض فقر الدم المنجلي، تم تحديد أحد عشر جينا: [ACKR1، AGER، FCER1A، HLA-A، HLA-DQB1، HLA-DRB1 HLA-G، NOTCH4، RRM1، STIM1، وTRIM5] قادرا على التفاعل مع ما مجموعه 114 مركبًا دوائيًا مختلفًا. بعض هذه الأدوية (عددها 36) لم تُعتمد بعد للاستخدام السريري، وبالتالي استبعدت من التجربة. جين (HBG2) لم يظهر تفاعلا مع أحد الأدوية المعتمدة. من بين الأدوية الـ 78 المتبقية، استبعد ما مجموعه 21 علاجًا كيمياويًا، كما استبعدت مادتان من الفيتامينات والمواد التشخيصية، وثلاثة لقاحات، و12 مضادًا ميكروبيًا. أما الأدوية المتبقية (عددها 40) فقد تم تصفية بعضها بناءً على عدم سلامتها.
نتيجةً لذلك، استبعدنا 5 من مضادات الصرع، و5 من مضادات الاكتئاب، و3 من مضادات الغدة الدرقية، والجلوكوكورتيكويدات، والتيكلوبيدين (دواء لمنع السكتات الدماغية)، والهيدرالازين (المستخدم لعلاج ارتفاع ضغط الدم) نظرًا لآثارها الجانبية غير المرغوب فيها. هيدروكسي يوريا (المستخدم لعلاج أمراض الدم) ومسكنات الألم (مثل، الأسيتامينوفين، والأسبرين، ولوميراكوكسيب) لم يتم تضمينها في القائمة النهائية المقترحة، لأن الأطباء يصفونها لمرضى فقر الدم المنجلي في عياداتهم بشكل روتيني.
ونتيجةً لذلك، قلصنا القائمة لتشمل التفاعلات بين 20 دواءً و6 جينات، حيث يتفاعل 18 من هذه الأدوية مع أربع جينات مضادات الكريات البيضاء البشرية HLA ( HLA-DQB1، وDRB1، وG، وA) ، يتفاعل الدواءان المتبقيان (أوماليزوماب “omalizumab” وأللوبيورينول “allopurinol”) مع جينين مختلفين (FCER1A وNOCH4، على التوالي) [كما هو موضح في الجدول رقم 1، للاطلاع على الجدول، راجع ج الورقة الأصلية].
الصنف العلاجي الأكثر شيوعًا من الأدوية المرشحة شملت مثبطات المناعة (عددها 12)، بما فيها: [أداليموماب adalimumab، وكاناكينوماب canakinumab، وإنفليكسيماب infliximab، وأوماليزوماب omalizumab، وتوسيليزوماب tocilizumab، وإيتانيرسيبت etanercept، وريلوناسيبت rilonacept، وبيغ إنتيرفيرون peginterferon ألفا-2أ و2ب، وآزاثيوبرين azathioprine، وغلاتيرامير glatiramer، وأناكينرا anakinra].
أما الصنف العلاجي الثاني الأكثر شيوعًا فهي الأدوية الخافضة للدهون (وعددها 7)، والتي شملت فينوفايبرات وستاتينات (بيتافاستاتين، وبرافاستاتين، وأتورفاستاتين، وسيمفاستاتين، وروزوفاستاتين، وفلوفاستاتين).
في نهاية المطاف، اقتُرحت مثبطات المناعة – بما فيها الأجسام المضادة وحيدة النسيلة – والستاتينات كأنسب الأدوية المرشحة لإعادة توظيفها لعلاج شدة مرض فقر الدم المنجلي، وذلك بناءً على تفاعلاتها مع الجينات الرئيسة الداخلة في المسارات المرضية الحرجة لمرض فقر الدم المنجلي، وبدعم من الأدلة المنشورة(18، 22). وتحديدًا، 3 مثبطات مناعة، يعمل كل منها على أهداف جينية مختلفة (أوماليزوماب يستهدف جين (FCER1A)، وكاناكينوماب وإيتانيرسيبت يستهدفان الجين (HLA-DRB1))، إلى جانب ستاتين (سيمفاستاتين، الذي يتفاعل مع كل من (HLA-G وHLA-DRB1).
هذه الأدوية المرشحة أظهرت درجة تفاعل عالية بين الدواء والجين. بالإضافة إلى ذلك، اقترحنا أللوبيورينول، وهو دواء مضاد لفرط زيادة حمض اليوريك في الدم، ويستهدف جين (NOTCH4)، باعتباره مرشحًا واعدًا لإعادة التوظيف.
4. مناقشة النتائج
برز تطبيق المعرفة الجينية لجعل إعادة توظيف الأدوية المعتمدة سابقًا استراتيجيةً واعدة ومبتكرة في مجال علم الأدوية. في عام 2021، كانت موافقة إدارة الغذاء والدواء الأمريكية (FDA) على ما يقرب من ثلثي الأدوية الجديدة مبنية على أفكار ودراسات مستمدة من علم الجينات الذي ساعد على اكتشافات دوائية مهمة(36، 37). تفيد التحليلات السابقة بأن الأدوية التي تستهدف الجينات المستندة إلى أدلة جينية بشرية رصينة تتمتع باحتمالية أعلى لتجاوز مرحلة التجارب السريرية والحصول على موافقة الهيئات التنظيمية للدواء والغذاء مقارنةً بالأدوية التي ليس لها هذا الدعم الجيني البشري(22).
ولكن الموافقة تتطلب أيضا تفاعلا معقدا بين عدة عوامل، بما في ذلك تصميم التجارب السريرية، وتنوع السكان (العرق والإثنية والثقافة واللغة والوضع السياسي والاقتصادي)، والاختيار الدقيق لنقاط النهاية (اختيار النتائج الموثوقة والقابلة للقياس والتي تصب في مصلحة المريض)، وبيانات سلامة الأدوية ومفعوليتها، بالإضافة إلى الخبرة المكتسبة بعد التسويق. بالرغم من أن الدعم الجيني يمكن أن يعزز الأساس البيولوجي والتحقق من صحة الهدف الكامن وراء تطوير الدواء، إلا أنه لا يضمن النجاح وحده. فالاحتمالية الإجمالية لموافقة الدوائر المختصة تعتمد في نهاية المطاف على اعتبار جميع هذه المعايير التنظيمية والعلمية والسريرية المتعددة الأوجه(38).
لقد أدى تطوير وتطبيق مناهج التحليل الجيني المتقدمة، مثل دراسات ترابط الجينوم الكامل (GWAS) وتسلسل الجيل التالي (NGS) مثل تسلسل كامل الجينوم (WGS) وتسلسل كامل الإكسوم (WES)، إلى تعميق فهمنا للأسس الجينية لسمات الإنسان المعقدة (طول القامة ولون الشعر ومستوى الذكاء، وقابلية الاصابة بأمراض معينة، مثلًا) بشكل كبير(39، 40, 41). وقد سهّل هذا التقدم تحديد أهداف دوائية واعدة، وذلك باكتشاف المتغيرات الجينية المرتبطة بالأعراض المختلفة لفقر الدم المنجلي(42).
ومع ذلك، ترجمة هذه الاكتشافات الجينية بفعالية إلى ممارسة سريرية ما تزال تواجه صعوبات، لا سيما في حالات مثل مرض فقر الدم المنجلي، الذي يتميز بسمات ظاهرية وأعراض متنوعة تتأثر بالارتباطات الجينية وعوامل بيئية متعددة (الأمراض والسمات الشائعة، مثل، الأمراض الناجمة عن مزيج معقد من الجينات المتعددة التي تتفاعل مع عوامل بيئية ونمط حياة مختلف، وليس جينًا واحدًا فقط). اعتبار استخدام الأدوية المعتمدة سابقًا يعد منهجيةً قيّمةً آخذةً في الازدياد والتوسع بالفعل في تطبيقات سريرية جديدة (علاجات جديدة) بتكلفة معقولة(30; 36، 43).
استخدمت العديد من الدراسات السابقة طريقة محاكاة حاسوبيةً لمعالجة وتحليل البيانات لاقتراح أدويةٍ يمكن أن تكون مرشحة وواعدة والتعرف على مركبات مستهدفةٍ لعلاج حالاتٍ طبيةٍ مختلفةٍ، وذلك بالاستفادة من نتائج تحليلات الترابط الجينومي الكامل المفتوحة لاستخدام العامة(30، 44، 45).
تتوفر قواعد مختلفة لبيانات الترابط الجينومي الكامل، ويمكن استخدامها لمعرفة الأساس الجيني لأمراضٍ بعينها. لذلك، من الممكن ترجمة إشارات معينة (أي جينات أو تغيرات بيولوجية) لتصميم أدوية لعلاج مرض معين استنادًا إلى علم الجينوم وخطةٍ بحثيةٍ مثبتة علميًا(30). في مشروعنا، اعتمدنا منهجيةً مماثلةً لاقتراح خياراتٍ علاجيةٍ دوائيةٍ جديدةٍ يُمكن أن تستهدف جيناتٍ محددةً معروفةً بتأثيرها الايجابي في مرضى فقر الدم المنجلي (أي تخفيف الأعراض ومضاعفات المرض وتحسين جودة الحياة وحتى المحافظة على البقاء على قيد الحياة).
في دراسة سابقة استخدمنا فيها الترابط الجينومي الكامل، اكتشفنا جينات متعددة لها دور هام في مرض فقر الدم المنجلي ومضاعفاته(18). علاوة على ذلك، تهدف الدراسة الحالية إلى تعظيم الاستفادة من البيانات المتاحة لترجمة المعرفة الجينية إلى رعاية سريرية (علاج دوائي) لمرضى فقر الدم المنجلي. وقد توصلت طريقتنا التي صممناها لتطوير استخدام جديد للعقاقير للتعرف على الجينات المحددة التي تعمل عليها بعض العلاجات المستخدمة فعليا لمرضى الأنيميا المنجلية مثل جين (RRM1) المؤثر في عمل دواء هيدروكسي يوريا، وجيني (HLA-DQB1) و(HLA-DRB1)، اللذين تعمل عليهما بعض المسكنات. هذه النتائج شكلت معايير للتحقق من صحة أداء خط تطوير العقاقير (المزيد من التفاصيل متوفرة في الملحق التكميلي). اكتشاف هذه الأدوية الموفق يشهد برصانة خط تطويرنا لهذه العلاجات.
استُبعدت هذه الأدوية العلاجية من التقييم الإضافي، نظرًا لكونها معتمدة بالفعل لعلاج مرض فقر الدم المنجلي، وليس بسبب مخاوف من كونها سمية. كان أحد الاعتبارات الرئيسة في خطتنا لإعادة استخدام الأدوية هو التقييم المنهجي لكل من الإمكانات العلاجية وخصائص السلامة للأدوية الثمانية والسبعين المرشحة لعلاج مرض فقر الدم المنجلي. طُبقت اعتبارات السلامة بدقة في كل خطوة من خطوات ترشيح كل دواء وتحديد أولويات الأدوية (من حيث كونها واعدة وفعالة وآمنة للعلاج).
بالإضافة إلى استبعاد دواء هيدروكسي يوريا، استُبعدت أدوية العلاج الكيماوي الأخرى أيضًا بناءً على علاقتها الموثقة بأعراض جانبية خطيرة، بما في ذلك سمية النخاع العظمي (أو كبت النخاع العظمي، والأورام الخبيثة الثانوية (أي الشخص المصاب بالفعل بسرطان قد يصاب بسرطان آخر مختلف)، وتسمم الكبد، وتسمم القلب، وكبت المناعة الشديد(33، 34). تُعد هذه المخاطر حرجة بشكل خاص على الفئات المريضة، مثل مرضى فقر الدم المنجلي.
جميع الأدوية المرشحة التي حددها خط تطوير العقاقير، بما فيها تلك التي استبعدت من خطوة التحليل الأولي (مثل الأدوية المستخدمة في العلاج الكيماوي)، يمكن الاطلاع عليها في الملحق لهذه الدراسة. هذا الملحق يُمكّن الأطباء والباحثين من تقييم جميع العوامل المقبولة من الناحية الميكانيكيًة بشفافية، وتصميم تجارب سريرية مُصممة خصيصًا لتقييم كل من الفوائد والمخاطر وفقًا لاحتياجات المريض الخاصة، والأدلة المستجدة، ومعايير الرعاية المتقدمة.
استبعاد بعض الأدوية لا يمنع من أخذها في الاعتبار مستقبلًا. يُذكر أن الدراسات الحديثة التي أُجريت على الأدوية المؤثرة على الصفات فوق الجينية (الجينات التي تؤثر منتجاتها مباشرة من خلال ميثلة الحمض النووي) والأدوية غير السامة للخلايا، أو منخفضة الجرعة أو تلك التي تؤخذ عن طريق الفم، مثل دواء ديسيتابين (decitabine)، بالإضافة إلى رباعي هيدرويوريدين (tetrahydrouridine) ، كانت نتائجها واعدة، حيث استُحث انتاج الهيموغلوبين الجنيني (HbF) بدون مشكلات، وحُسّنت مقاييس مرض فقر الدم المنجلي (تلك المستخدمة لتتبع صحة المريض، وشدّة المرض، وفعالية العلاج، وجودة الرعاية لمرض فقر الدم المنجلي، بما في ذلك مستويات الهيموغلوبين، وغيرها من المضاعفات) دون أن تسبب تثبيطًا معتبرًا لنخاع العظم (لإنتاج خلايا الدم الحمراء والبيضاء) أو مخاطر العلاج الكيمياوي التقليدية (مثلًا، عدم تمييز العلاج بين الخلايا المصابة والصحيحة)(46). تشير هذه التطورات إلى أنه ينبغي إبقاء أدوية العلاج الكيمياوي المعينة، عند تحسين سلامتها، في الاعتبار باعتبارها جانبًا من المشهد العلاجي الأوسع لمرض فقر الدم المنجلي – خاصةً في الحالات المُقاومة للعلاج أو الشديدة.
قُلصت القائمة المرشحة إلى عشرين دواءً معتمدًا، وذلك لقدرتها القوية على التفاعل مع ست جينات تُعدّل أنماطًا ظاهريةً مُختلفة لمرض فقر الدم المنجلي (وهي الجينات التي تغير أو تعدل أعراض المرض في مختلف المرضى). تُصنّف الأدوية المُرشّحة المُختارة إلى أدوية مُعدّلة للدهون (تنظم مستويات الدهون) وأخرى مُعدّلة للمناعة (مثبطة للمناعة في هذه الحالة)، مع إمكانية تعديل مسارات مُتعددة في مرض فقر الدم المنجلي، بما في ذلك وظيفة بطانة الأوعية الدموية، وانحلال الدم، وتمنجل خلايا الدم(47). بناءً على معايير فريدة.
نتج من عملية التصفية هذه خمسة أدوية واعدة جدًا ومرشحة لإعادة الاستخدام لعلاج فقر الدم المنجلي، وهي: [سيمفاستاتين simvastatin وأللوبورينولallopurinol وكاناكينوماب canakinumab وإيتانيرسيبت etanercept] – وجميعها سبق أن خضعت لتجارب سريرية صغيرة على مرضى فقر الدم المنجلي – بالإضافة إلى أوماليزوماب (omalizumab). إضافة إلى سيمفاستاتين، والذي ينتمي إلى مجموعة أدوية ستاتين، تم التعرف على أدوية أخرى من نفس المجموعة أيضًا، مثل أتورفاستاتين، وبرافاستاتين، وفلوفاستاتين، وروزوفاستاتين، وبيتافاستاتين، والتي كانت نتائجها أيضًا واعدة في إدارة فقر الدم المنجلي، نظرًا لخصائصها المُضادة للالتهابات.
تُخفِّض هذه الأدوية عامل النسخ (NF-KB) (العامل النووي المعزز لسلسلة كابا (Kaba) الخفيفة في الخلايا البائية النشطة “NF-κB”) وتُثبِّط انتاج السايتوكينات الرئيسة المُسبِّبة للالتهابات مثل (TNF) و(IL1B) ، وتُحسِّن وظيفة بطانة الأوعية الدموية من خلال استعادة التوافر البيولوجي لأكسيد النيتريك (NO) وتقلل جزيئات الالتصاق (للحد من التصاق خلايا الدم المنجلية)، مما تُسهم مجتمعةً في التأثير الوقائي للأوعية الدموية(48، 49). والجدير بالذكر أن تجربة جوبيتر (JUPITER) (الأحرف الآولى لاسم التجربة: تبرير استخدام الستاتينات في الوقاية الأولية: تجربة تدخلية لتقييم تجربة دواء روزوفاستاتين)، أثبتت قدرة الستاتينات المُعَدِّلة للمناعة، حيث خفَّض دواء الروزوفاستاتين مستويات البروتين التفاعلي (-C) ويرمز له بـ (CRP ) (بروتين تنتجه المعدة ويرتفع مستواه في الدم في حالات الاصابات والالتهابات، ويعد علامة حيوية على ذلك) بشكل ملحوظ بنسبة تقارب 37%، مما يُعزِّز دور الستاتينات في التخفيف من المضاعفات المرتبطة بالالتهابات(50).
اقتُرح دواء سيمفاستاتين باعتباره مرشحًا مثاليًا من بين جميع الستاتينات لتفوقه في التفاعل وأهميته السريرية. بينما تتفاعل أدوية الستاتينات الأخرى مع جين (HLA-DRB1) فقط، الذي يُشفّر بروتينًا معقداً له دور في التوافق النسيجي الرئيس عند زراعة الأعضاء، يتفاعل سيمفاستاتين بشكل فريد مع كلٍّ من جين (HLA-DRB1) وجين (HLA-G) ويُعدّ هذان البروتينان أساسيان في الاستجابات المناعية التي تُفاقم مضاعفات فقر الدم المنجلي، مثل نوبات الانسداد الوعائي وتلف الأعضاء(51 – 53). علاوة على ذلك، أثبت دواء سيمفاستاتين قدرة أعلى على خفض مستويات عامل ڤون ويلبراند (vWF) (سلسلة من بروتين سكري متعدد، ويعد عامل تخثر)، مما يُؤكد دوره الحاسم في إدارة فرط التخثر المرتبط بمرض فقر الدم المنجلية(54، 55).
أثبتت دراسة سريرية أُجريت على مجموعة صغيرة من مرضى فقر الدم المنجلي (عددهم 26) أن تناول دواء سيمفاستاتين في المدى القصير حسّن بشكل ملحوظ التوافر البيولوجي لأكسيد النيتريك بنسبة 52% تقريبًا (القيمة الاحتمالية = 0.01، أي أن هناك احتمال مقداره 1٪ بأن النتيجة التي حصل عليها الباحثون حدثت بالصدفة أو بشكل عشوائي، وهذا يعتبر دليلًا قويًا غير مباشر على صحة النتيجة) وانخفضت المؤشرات البيولوجية الالتهابية الجهازية مثل البروتين التفاعلي-C (وIL-6 (إنترلوكين – 6)، وكلاهما يعتبران علامتان حيويتان على الالتهاب ومرتبطان باحتمال زيادة ضعف الأوعية الدموية في مرضى فقر الدم المنجلي(56). علاوة على ذلك، أفادت دراسة تجريبية أُجريت على 19 مريض فقر دم منجلي أن دواء سيمفاستاتين قلل من حدوث ألم الانسداد الوعائي بنسبة 85% (القيمة الاحتمالية = 0.0003، مما يدل على صحة النتيجة وفاعلية الدواء) وقلّل من استخدام المسكنات بنسبة 67% (القيمة الاحتمالية = 0.003).
كما كان هناك انخفاض ملحوظ في البروتين التفاعلي-C في الدورة الدموية، وسيليكتين (E) الذائب (s) وجزيء الالتصاق بين الخلايا 1 (sICAM-1) وجزيء الالتصاق بين الخلايا 3 (sICAM-3) وعامل نمو بطانة الأوعية الدموية، مع ملاحظة التأثيرات الأكثر وضوحًا لدى المرضى الذين يأخذون دواء هيدروكسي يوريا بالتزامن(56). بالرغم من أن الدراسات المذكورة حول الستاتينات في مرض فقر الدم المنجلي تُشير إلى انخفاض واعد في مؤشرات الالتهابات البيولوجية ونوبات الألم السريرية، إلا أن أحجام العينات في تلك الدراسات كانت متواضعة (حيث بلغ عدد الأفراد بين 19 و26 مريضا فقط)، مما يؤثر سلبًا في رصانة النتائج وإمكانية تعميمها. لذلك، ينبغي اعتبار الأدلة الحالية أولية، ما يبرر الحاجة إلى تجارب عشوائية منضبطة بحجم عينة عالٍ (عدد أشخاص كافي) لمعرفة التأثيرات المستقلة لعلاج مرض فقر الدم المنجلي بالستاتينات بشكل أوضح.
الدراسات المخبرية (خارج الجسم) إمكانات سيمفاستاتين العلاجية، حيث تُبين أن زيادةً بمقدار 1.9 ضعف في انتاج الهيموغلوبين الجنيني وانخفاضًا بنسبة 30%-35% في حالات الخلايا المنجلية التي تبقى متمنجلة ولا يمكن إصلاحها في ظل نقص الأكسجين(57). بناءً على هذه الأدلة الرصينة والمعايير المحددة، نوصي بدراسة إكلينيكية لتأكيد فوائد استخدام دواء سيمفاستاتين باعتباره دواءً مثاليًا مقترحا في علاج حالات داء فقر الدم المنجلي، مع أن أدوية الستاتينات الأخرى قد يكون لها فوائد أيضًا، وذلك بحسب حالة المريض. يتطلب الأمر إجراء المزيد من الدراسات، بما فيها التجارب السريرية المعشاة واسعة النطاق وطويلة المدى، لتوضيح الإمكانات العلاجية لدواء سيمفاستاتين بشكل كامل في هذا السياق.
[كلمة ‘معشاة’ الواردة في الجملة السابقة تعني ما يلي: تُبرز دراسة مصممة لتقييم فعالية أو سلامة التدخل العلاجي، وذلك بالتوزيع العشوائي للمشاركين على مجموعة مقارنة واحدة أو أكثر لاستبعاد أي نوع من التحيز. في هذا التصميم، تتلقى إحدى المجموعات التدخل العلاجي (دواء) قيد الدراسة، بينما تتلقى المجموعة الأخرى علاجًا بديلًا، أو دواءً وهميًا].
تفيد الأدلة السريرية المتراكمة بأن أدوية الستاتينات (مثل دوائي سيمفاستاتين وأتورفاستاتين) أن لها فوائد معتبرة باعتبارها علاجًا لداء فقر الدم المنجلي عمومًا، بأعراض جانبية خفيفة، ومتوقعة، وقابلة للإدارة، والتحمل. علاوة على ذلك، تتوافر أدوية الستاتينات على نطاق واسع، وبأسعار معقولة، ومناسبة للعلاج المنزلي طويل الأمد، حيث أن منافعها تفوق كثيرًا مضارها على الأمد الطويل(56). من الأمور التي ينبغي ملاحظتها هي أن الاعتلال العضلي الناتج عن الستاتينات، بالرغم من ندرته عمومًا، قد يكون له أهمية متزايدة في فقر الدم المنجلي، نظرًا لارتفاع معدل انتشار الأمراض الكلوية أو الكبدية المصاحبة لفقر الدم المنجلي(58).
كما أن التداخل مع أعراض الجهاز العضلي الهيكلي لداء فقر الدم المنجلي قد يعيق الكشف المبكر عن سمية هذه الأدوية. بالرغم من أن الدراسات التجريبية الحالية لم تلاحظ اعتلالًا عضليًا زائدًا، إلا أن أحجام العينات ومدة المتابعة كانت محدودة(56). لذلك، نوصي بالمراقبة الروتينية لمستويات إنزيم كاينيز الكرياتين (creatine kinase) في الدم (للكشف عن احتمال اصابات قذ تؤدي إلى اعتلال عضلي أو إضرار بالقلب) والنظر في الاختبارات الصيدلانيّة الجينية (على سبيل المثال، النمط الجيني (SLCO1B1) وهو جين يعطي الكبد تعليمات لتخليق بروتين مسؤول عن تنظيف الجسم من الهرمونات والأدوية، وخاصة أدوية الستاتينات) للتخفيف من المخاطر وتخصيص العلاج للشخص المناسب(59).
من المقترحات الأخرى لإعادة الاستخدام دواء أللوبيورينول، الذي يستهدف نواتج جين (NOTCH4) وينتمي هذا الجين إلى عائلة (NOTCH) وهو معروف بدوره في تنظيم وظيفة بطانة الأوعية الدموية وله تأثيرات مضادة للالتهابات، بالإضافة إلى تأثيره في تكوِّن خلايا الدم وصفائح الدم، وكل ذلك قد يُسهم في تخفيف شدة المرض(60، 61). في مجموعة بيانات مرضى فقر الدم المنجلي السعوديين، تعرفنا على المتغير الجيني رقم (rs3132946). وتشير المعلومات إلى تعدد أشكال النيوكليوتيدات المفردة المقترنة بالعديد من الأمراض المناعية والالتهابية في جين (NOTCH4). المتغير (rs3132946) سبق اكتشافه باعتباره علامة حيوية مرتبطة بأمراض الرئة الخلالية (ILD ) (أمراض تصيب النسيج الخلالي الرئوي)(62).
تُعد هذه الحالة شائعة بشكل ملحوظ لدى مرضى فقر الدم المنجلي، حيث ظهر على 74٪ من الراشدين في دراسة حشدية (ويطلق عليها دراسة التعرض، وتتميز بأنها تحتاج عينة كبيرة وأنها مستقبلية وطويلة الأمد) لمرضى ذوي نمط تقييدي في وظائف الرئة (حيث لا تستطيع الرئتان الامتلاء بالهواء نظرًا لحجمهما الصغير)(63). بالرغم من أن الأعراض المشتركة لأمراض الرئة المرتبطة بفقر الدم المنجلي (أي تلك التي هي من مضاعفات فقر الدم المنجلي) لا تظهر على كل الراشدين المصابين بمرض فقر الدم المنجلي، إلا أن هذا النمط التقييدي يبقى مستمرًا في الظهور سريريًا بنفس الطريقة وعلى نفس المنوال، مما يؤكد تأثيره المعتبر إحصائيًا في المجموعة المصابة بفقر الدم المنجلي.
دواء أللوبيورينول، وهو مثبط إنزيم أوكسيديز الزانثين المعروف (أي يخفض نشاطه)، والذي استُخدم لفترة طويلة لعلاج فرط حمض يوريك الدم لدى مرضى النقرس أو متلازمة انحلال الورم (تحدث عند تدمير الأورام السرطانية بسرعة وانتشار محتوياتها في الدم)(64)، أظهر فوائد وعائية أوسع. إذ يمكنه حماية الأنسجة الوعائية من الإجهاد التأكسدي وإصابات إعادة التروية المتكررة (يحدث عندما يتدفق الدم مرة أخرى بعد نقص التروية أو نقص الأكسجين)، وكلاهما عنصران أساسيان في أمراض فقر الدم المنجلي(65). وبتثبيط إنتاج أنواع الأكسجين التفاعلي (ROS) يحافظ دواء أللوبيورينول على التوافر البيولوجي لأكسيد النيتريك، مما يدعم استرخاء الأوعية الدموية ووظيفتها(66).
تُظهر الدراسات ما قبل السريرية على نماذج حيوانية مصابة بفقر الدم المنجلي نتائج متباينة؛ فبينما لم تُشر إحدى الدراسات إلى أي تأثير يُذكر على التصاق الخلايا (إما ببعضها، أو بسطح، أو غشاء)، أظهرت دراسة أخرى تحسنًا في تدفق الدم وانخفاضًا في تحشيد كريات الدم البيضاء عند التعرض إلى (العلاج بـ) دواء أللوبيورينول(67؛ 68). نظرًا للإجهاد التأكسدي وخلل وظائف البطانة الوعائية الملحوظ في مرض فقر الدم المنجلي، فإن إجراء المزيد من الدراسات حول الإمكانات العلاجية لدواء أللوبيورينول، وخاصةً عند استخدامه مع أدوية أخرى، أمرٌ ضروري. ومع ذلك، فإن الدراسة الدقيقة لآثاره (أعراضه) الجانبية، بما في ذلك فرط الحساسية وردود الفعل الشديدة النادرة، أمرٌ بالغ الأهمية. حتى في المجتمعات التي لا ينتشر فيها بشكل كبير طفرة (HLA-B*5801)، وهو مضاد من مضادات الكريات البيضاء البشرية (HLA) المهمة في علم الوراثة المناعية، ترتبط هذه الطفرة بشكل خاص بزيادة احتمال تفاعلات فرط الحساسية لدواء أللوبيورينول الشديدة(69). تحديد المكون الجيني لدواء أللوبيورينول من خلال معرفة الطفرة في جين (HLA-B*5801) قد يساعد في التخفيف من خطر تفاعلات فرط الحساسية بسبب دواء أللوبيورينول عند وصفه للمريض(70).
بالرغم من أن الأجسام المضادة وحيدة النسيلة والأدوية ذات العلاقة (مثل أدوية أوماليزوماب، وكاناكينوماب، وإيتانيرسيبت) تتطلب حُقنًا منتظمة وتكلفتها المالية عالية، إلا أنها أثبتت سلامتها وفعاليتها في خفض الالتهاب والمضاعفات الحادة في مرض فقر الدم المنجلي. أما الآثار الجانبية المعروفة، مثل ارتفاع احتمالية الاصابة بالعدوى، فهذه الأثار يمكن إدارتها بشكل عام، ونفع هذه الأدوية في قدرتها على استهداف الأعراض الحادة لمرض فقر الدم المنجلي في حالات معينة تعتبر أعلى من ضررها بكثير(71).
وقد تبين بعد استخدامها أن سلامتها مقبولة في المراكز الصحية الحديثة التي يقدم فيها التشخيص والعلاج، مثل المستشفيات والعيادات، لمرض فقر الدم المنجلي وحالات فرط الالتهاب الأخرى، دون ملاحظة أي زيادة في الأعراض الجانبية الخطيرة(72). وقد اعتمدت إدارة الغذاء والدواء الأمريكية (FDA) دواء أوماليزوماب في البداية عام 2003 لعلاج الربو المزمن الذي يتراوح في الشدة من المتوسط إلى الحاد، ثم بعد ذلك وسعت نطاق استخدامه في السنوات اللاحقة(73). وقد برز هذا الدواء، الذي يستهدف نواتج جين (FCER1A)، باعتباره أحد أبرز الأدوية المرشحة لإعادة الاستخدام. هذا الغلوبيولين المناعي المُؤتلف G (IgG) هو جسم مضاد وحيد النسيلة يرتبط انتقائيًا بـ الغلوبيولين المناعي (IgE) الحر (غير المؤتلف)، ما من شأنه أن يُخفف من حدة الربو التحسسي.
وذلك بمنع (IgE) من التفاعل مع مستقبلات (Fcε) عالية الألفة (المسؤولة عن التوسط في التفاعلات التحسسية وذلك بالارتباط بمنطقة الـ (Fc) من الجسم المضاد للغلوبيولين المناعي E (IgE) على الخلايا المستفحلة (التي تستجيب إلى مثير ما)، مثل الخلايا البدينة (خلايا مناعية توجد في النسيج الضام). يُعدل دواء أوماليزوماب التفاعلات المثيرة للالتهابات التي تُؤدي إلى التهاب قصبة الهواء، وقد يُحد أيضًا من اضطرابات التخثر المرتبطة بنشاط مرض فقر الدم المنجلي(74).
في مجموعة مرضى فقر الدم المنجلي السعوديين، تم الكشف عن متغير (rs2494250) في جين (FCER1A)، وقد ذكر سابقًا اقتران هذا المتغير بشكل معتبر احصائيًا بارتفاع مستويات المؤشرات الحيوية الالتهابية، بما فيها (CCL2/MCP-1) (ربيطة الكيموكين 2 / بروتين الجذب الكيمائي للخلايا الوحيدة-1، ويعتبر عاملًا مهمًا في تحشيد الخلايا المناعية، لمواقع الالتهاب)(75). علاوة على ذلك، أفادت دراسات بارتفاع معدل انتشار (IgE) بين السعوديين المصابين بفقر الدم المنجلي، مما قد يزيد من احتمال إصابتهم بمتلازمة الصدر الحادة (ACS) والتي منها المتلازمة التاجية الحادة ومتلازمة مخرج الصدر ومتلازمة المسكة البركية). تجدر الإشارة إلى أن متلازمة الصدر الحادة مسؤولة عن 25% من حالات دخول المرضى وحدات العناية المركزة و28.5% من الوفيات بين مرضى داء فقر الدم المنجلي، وبتسجيل معدلات مرتفعة مصابة بهذه الحالات بشكل خاص لدى أفراد من منطقة الأحساء(76 – 78). يلزم إجراء المزيد من الدراسات السريرية على مرضى فقر الدم المنجلي للتأكد من فوائد دواء أوماليزوماب في الحد من مضاعفات هذا المرض.
علاوة على ذلك، خضع دواء كاناكينوماب (canakinumab) للدراسة على 49 طفلاً وشاباً مصاباً بفقر الدم المنجلي في دراسة حديثة مزدوجة التعمية ومعشاة(12، 72). وقد أثبت الدواء مستوى أمان مقبولاً، بعكس أدوية الأجسام المضادة وحيدة النسيلة الأخرى التي غالباً ما تقترن باحتمال الإصابة بالعدوى(80، 81). ورغم أن التجربة المعشاة والمقارنة بدواء وهمي (بلاسيبو) لدواء كاناكينوماب على الأطفال والشباب المصابين بفقر الدم المنجلي لم تحقق هدفها الرئيس المتمثل في خفض حدة الألم اليومي مقارنةً بالحالة بدون علاج، فقد أثبتت العديد من النتائج الثانوية فوائد ممكنة. وعلى وجه التحديد، انخفاض مستويات المؤشرات الحيوية الالتهابية، وانخفاض معدلات دخول المستشفى، وتحسن في النتائج التي أبلغ عنها المرضى ذاتيًا، مثل مستوى التعب والإرهاق والقدرة على الدراسة أو العمل (عدم التغيب عن العمل أو الدراسة).
تشير هذه النتائج إلى أنه بالرغم من عدم تحقيق هدف العلاج المتمثل في خفض حدة للألم، إلا أن دواء كاناكينوماب قد يكون له فوائد أخرى في إدارة جوانب أخرى من مرض فقر الدم المنجلي، ولكن هناك حاجة إلى مزيد من الدراسات لتأكيد جدواه من الناحية السريرية (مدى فائدة الفحص أو التدخل العلاجي في تحسين صحة المريض، والاسترشاد بالمعلومات لاتخاذ القرارات العلاجية، وتحسين الرعاية الصحية، أو إدارة أفضل للمرض). من جهة أخرى، دواء إيتانيرسيبت (etanercept)، وهو دواء مُثبِّط لمستقبلات عامل نخر الورم ألفا (TNF-α) قد يُوقف المؤشرات الالتهابية (المثيرة للجهاز المناعي) التي يبدأها عامل نخر الورم ألفا المسبب للالتهاب(78). ويمكن تقليل نوبات إصابة نقص التروية وإعادة التروية، التي تؤدي عادةً إلى خلل في وظائف الأوعية الدموية وما يتبعه من انسداد وعائي في مرض فقر الدم المنجلي، باستخدام دواء إيتانيرسيبت(28).
الاستخدام طويل الأمد لدواء إيتانيرسيبت لعلاج فئران مصابة بمرض فقر الدم المنجلي قد يؤدي إلى انخفاض في أعراض المرض الحادة، بما فيها الانسداد الوعائي، والاستجابات لمثيرات الألم، وعدد الكريات البيضاء، والمؤشرات الحيوية الالتهابية(82). بالإضافة إلى ذلك، تم علاج حالتين سريريتين لمرضى مصابين بالتهاب المفاصل الروماتويدي وفقر الدم المنجلي بنجاح وأمان بدواء إيتانيرسيبت(83). هذه الأدلة تدعم الحاجة إلى إجراء تجارب سريرية لتقييم نتائج دواء إيتانيرسيبت لدى مرضى فقر الدم المنجلي.
لهذه الأدوية المقترحة إمكانات واعدة في إدارة حدة مرض فقر الدم المنجلي، وتتميز بخصائص متنوعة، حيث تعمل بطرق مختلفة ويمكن تناولها بأشكال مختلفة (حبوب أو حقن). كما أن بعضها يتميز بانخفاض تكلفتها، ما من شأنه أن يسهل اعتمادها سريريًا واستخدامها من قبل الكادر الصحي. على سبيل المثال، يُستخدم دواء سيمفاستاتين (simvastatin) ودواء أللوبيورينول (allopurinol) على نطاق واسع، وهما متوفران في تركيبات فموية (سائل أو حبوب)، ويمكن وصف جنيس هذه الأدوية للمرضى (جنيس الدواء هو دواء مكافئ ومطابق للدواء الأصلي) وتباع بأسعار معقولة، مما يسهل إمكانية الحصول عليهما للاستخدام طويل الأمد(76، 83، 84).
في المقابل، تؤثر الأجسام المضادة وحيدة النسيلة أيضًا في مسارات حدة المرض (تقلل مدى حدته)؛ بيد أنها تتطلب حقنًا تحت الجلد، تُعطى غالبًا أثناء زيارات المريض المستشفى بين كل 4-8 أسابيع، ما يجعلها غير مريحة أو ملائمة للمريض. بالإضافة إلى ذلك، قد تُحدّ تكاليفها المرتفعة مقارنةً بالأدوية الفموية من إمكانية الحصول عليها(78، 85، 86). وبالتالي، الأدوية الفموية تعتبر أكثر ملاءمة حيث يُمكن تناولها يوميًا في البيت، مما قد يُحسّن من التزام المريض بالعلاج، خاصةً في حالات الأمراض المزمنة، مثل فقر الدم المنجلي.
رغم أن العلاجات البيولوجية التي يمكن حقنها قد تكون أكثر فعالية، إلا أنها لا تخلو من تحديات تتعلق باضطرار المريض على التردد على مقدمي الرعاية الصحية في العيادات والمستشفيات لتلقي حقن الدواء، وارتفاع التكاليف. أما الأدوية الفموية فقد يكون استخدامها خيارًا استراتيجيًا قيّما لعلاج فقر الدم وإدارة مضاعفاته بناءً على أفضلية التزام المريض المتوقعة واعتبارات مكان تلقي الرعاية الصحية.
إحدى الطرق المهمة لإحداث تأثير إيجابي في مرض فقر الدم المنجلي وتقليل تفاعلاته في جسم المصاب هي استهداف الآليات المرضية المتعددة الممكنة. لذا، فإن اقتراح استخدام أدوية متعددة ذات ميكانيكية عمل مختلفة لمعالجة المرض مثل تعديل مستوى الهيموغلوبين الجنيني، ومنع أو تقليل التصاق خلايا الدم ببعضها وتمنجلها، والالتهاب، ونقص التروية ومخاطر إعادة التروية، والإجهاد التأكسدي، وتخثر أو تجلط الدم، وسمية الهيم الحر (الذي يسمم الأنسجة والأعضاء ويؤثر سلبًا في وظائف الكلى، والخلايا العصبية، وخلايا القلب، وخلايا الكبد، وكريات الدم البيضاء المحيطية). ولتحقيق ذلك، يُوصى باستخدام مزيج من علاجات دوائية لها آليات مختلفة(28).
في سياق استكشاف أهداف دوائية جديدة واعدة، نجحنا في التعرف على مواضع قابلة للاستهداف (أي المواضع التي يمكن أن يرتبط بها الدواء ويغير وظيفتها) في الكثير من منتجات الجينات التي لم تكن مستهدفة سابقًا ضمن هذه الدراسة. نوصي بشدة باستهداف مجموعة جينات مستقبلات شمية (في الأنف) باعتبارها أهدافًا جديدة، ولا سيما جينات [OR51V1 وOR52A1 وOR51B5]. الطفرات المهمة في هذه الجينات – مثل (rs7933549) (طفرة مغلطة، وهي طفرة نقطية في نيكولتيدة واحدة)، و (rs112098990) (طفرة إزاحة الإطار)، و (rs147062602) (طفرة إزاحة الإطار)، على التوالي – هي جينات شائعة لدى الأفراد المصابين بمرض فقر الدم المنجلي وترتبط بعيوب في وظيفة البروتين وطريقة تحلله لاحقا.
أفاد خطنا لتطوير الأدوية أيضًا بأن بروتينات [TRIM 6 وSIDT2 وCADM3] حيث تُعد أهدافًا علاجية جديدة وواعدة. بروتينات عائلة [TRIM، بما فيها TRIM6 وTRIM22 وTRIM34]، معنية بعلاج المناعة الطبيعية (لتعزيز الوقاية من مسببات الأمراض، والسيطرة على الأمراض، أو منع الاستجابات المناعية غير المرغوب فيها)، وتقدم دورة الخلية (من تطور وانقسام)، وتنظيم النسخ الجيني (الآليات التي تستخدمها الخلية في تنظيم تحول الدنا إلى رنا، وبالتالي تنسيق النشاط الجيني)، مما يوحي بأنها قد تؤثر في الالتهاب، والإجهاد التأكسدي، وتمايز الخلايا الجذعية المكونة للدم، والاستجابات المناعية(88). وتُعد هذه العمليات بالغة الأهمية في الفيسيولوجيا المرضية لمرض فقر الدم المنجلي ومضاعفاته.
علاوة على ذلك، قد يُساهم بروتين جين (SIDT2) في مضاعفات مرض فقر الدم المنجلي، إذ يرتبط باختلال تنظيم بروتين جين (SIDT2) بالكثير من الخصائص الأيضية (تحويل الغذاء إلى طاقة) المرتبطة بأمراض الكبد، واضطرابات القلب والأوعية الدموية، واعتلال الكلى المصاحب لفقر الدم المنجلي(89، 90). يلعب جين (SIDT2) دورًا في وظيفة إنزيمات اللايسوسوم (lysosomes) ، وقد يؤدي تعطيلها إلى حدوث خلل في معالجة النفايات الخلوية واختلالات في العملية الأيضية، مما يُفاقم اختلال وظائف الأعضاء وحصول بعض المضاعفات الشائعة لدى مرضى فقر الدم المنجلي. في الوقت نفسه، قد يُؤدي اختلال تنظيم بروتين جين (CADM3)، المعني بتكاثر الخلايا الظهارية (زيادتها في البول قد يشير إلى حالات التهابية وأمراض الكبد والكلى) والتصاق الخلايا ببعضها في دورة الخلية(91)، إلى إضعاف التكامل المناسب لنخاع العظم وتعطيل عملية تكوين الدم الطبيعي، مما قد يُؤثر في إنتاج خلايا الدم الحمراء وغيرها من خلايا الدم ونضوجها.
قد تؤدي هذه التغيرات في عمليات تكوين الدم إلى تفاقم الأنيميا، أو زيادة عدد الخلايا غير الناضجة في الدورة الدموية، أو تثبيط أو تعزيز الاستجابات المناعية، وكلها عوامل حاسمة في تباين وحدّة الأعراض السريرية لمرض فقر الدم المنجلي(92). يتمتع كل هدف من هذه الأهداف بدرجة عالية من قابلية الاستهداف الدوائي (مؤشر قابلية الاستهداف الدوائي هو مقياس حوسبي يتراوح عادةً ما بين 0 و1، ويتنبأ بمدى سهولة استهداف موضع ارتباط الدواء بالبروتين)، تتراوح بين 0.7 و0.86، ما يجعلها مرشحة بقوة لتطوير أدوية في المستقبل.
4.1 قيود النتائج
بالرغم من أن الدراسة الحالية قدمت إطارًا واعدًا لإعادة استخدام الأدوية المعتمدة، كما تعرفنا من خلالها على أهداف دوائية جديدة لإدارة حدة فقر الدم المنجلي، إلا أنها لا تخلو من بعض القيود (محدوديات) التي تحد من بعض صلاحية نتائجها. من تلك القيود هي أن الدراسة اعتمدت في منهجيتها جزئيًا على التكنولوجيا الحوسبية وعلم تحليلات المعلوماتية الحيوية (علم الأحياء الحوسبي). ومع أن هذين النهجين يملكان قوة تنبؤية إلّا أنهما غير قادرين على استيعاب كل التعقيدات التي تنطوي عليها التفاعلات بين الدواء والجينات في تجارب خارج الجسم الحي. إلا أن هذا الأمر تمت معالجته جزئيًا من خلال إجراء تجارب سريرية سابقة على حيوانات، ولكن عينة التجارب كانت صغيرة الحجم، والتي، ولله الحمد، اتسقت مع نتائج دراستنا، إلًا أننا لا نزال بحاجة إلى دراسة مستقبلية طولية وأكبر حجمًا.
للاطلاع الكامل على القيود الأخرى راجع الفقرة 4.1 في الورقة الأصلية.
4.2 الاستنتاجات
تشير التحليلات السابقة إلى أن احتمالية حصول الأدوية، التي تستهدف جينات مدعومة بأدلة جينية بشرية قوية، مثل نتائج دراسات الارتباط على مستوى الجينوم الكامل (GWAS)، على موافقة سريرية تعتبر أفضل مقارنةً بتلك التي تفتقر إلى مثل هذه الأدلة. بيد أن احتمالية الموافقة الإجمالية تعتمد على عدة عوامل أخرى، لا على الأدلة الجينية وحدها. وباستخدام منهجية راسخة تمكنا من التعرف على العديد من الأدوية المرشحة والواعدة لإعادة الاستخدام لعلاج حدة مرض فقر الدم المنجلي، ولا سيما أدوية الستاتينات، وأدوية تعديل المناعة، ودواء الأللوبيورينول (allopurinol).
بالإضافة إلى ذلك، تعرفنا على أهداف جديدة – وتحديدًا على عائلتي مستقبلات الجينات الشمية (OR) وجينات [(tripartite motif (TRIM] – لتطوير أدوية ممكنة للتخفيف من حدة مرض فقر الدم المنجلي ومضاعفاته، مع التركيز على السكان السعوديين. وقد استند اختيار هذه الأهداف الجينية إلى حصولها على درجات عالية لقابلية الاستهداف دوائيًا وانخراطها في المسارات الجزيئية الرئيسية الكامنة وراء المرض. لا تُعزز هذه النتائج فهمنا للأساس الجيني لمرض فقر الدم المنجلي فحسب، بل تُمهد الطريق أيضًا إلى مقاربات علاجية أكثر استهدافًا ومفعولية.
تبدأ خارطة الطريق المنطقية لترجمة هذه النتائج بدراسات مخبرية مُوجَّهة لتأكيد التأثير الوظيفي للتفاعلات الدوائية الجينية المُحدَّدة والأهداف الجديدة، تليها دراسات حيوية (داخل الجسم الحي) لتقييم الفعالية العلاجية والسلامة (الأمان) عند مرضى الأنيميا المنجلية تحديدًا. بعد ذلك، يمكن الانتقال بالأدوية المرشحة والواعدة إلى تجارب سريرية في مراحلها المبكرة لتقييم الجدوى السريرية، والجرعات المثلى، واستجابة المرضى، مما يمهد الطريق لعلاجات مُوجَّهة جينيًا ومُخصَّصة لفئات مُحدَّدة من مرضى فقر الدم المنجلي. تُعدّ التجارب السريرية المستقبلية وطويلة الأمد ضرورية للتحقق من صحة هذه التنبؤات الحوسبية وترجمتها إلى علاجات قابلة للتطبيق من شأنها أن تُحسِّن من مخرجات مرضى فقر الدم المنجلي.
الهوامش:
1- Inusa, B. P. D., Hsu, L. L., Kohli, N., Patel, A., Ominu-Evbota, K., Anie, K., et al. (2019). Sickle cell disease-genetics, pathophysiology, clinical presentation and treatment. Int. J. Neonatal Screen. 5 (2), 20. doi:10.3390/ijns5020020
PubMed Abstract | CrossRef Full Text | Google Scholar
2- Belisário, A. R., Silva, C. M., Velloso-Rodrigues, C., and Viana, M. B. (2018). Genetic, laboratory and clinical risk factors in the development of overt ischemic stroke in children with sickle cell disease. Hematol. Transfus. Cell Ther. 40 (2), 166–181. doi:10.1016/j.bjhh.2017.08.008
3- Nnodu, O. E., Oron, A. P., Sopekan, A., Akaba, G. O., Piel, F. B., and Chao, D. L. (2021). Child mortality from sickle cell disease in Nigeria: a model-estimated, population-level analysis of data from the 2018 demographic and health survey. Lancet Haematol. 8 (10), e723–e731. doi:10.1016/S2352-3026(21)00216-7
PubMed Abstract | CrossRef Full Text | Google Scholar
4- GBD 2021
5- Sickle Cell Disease Collaborators McHugh, T. A., Oron, A. P., Teply, C., Lonberg, N., Vilchis Tella, V., Kassebaum, N. J., et al. (2023). Global, regional, and national prevalence and mortality burden of sickle cell disease, 2000-2021: a systematic analysis from the global burden of disease study 2021. Lancet Haematol. 10 (8), e585–e599. doi:10.1016/S2352-3026(23)00118-7
PubMed Abstract | CrossRef Full Text | Google Scholar
6- Kumar, A., and Bhattacharya, S. (2024). Sickle cell disease: a comparative perspective on global and national initiatives. Front. Hematol. 3, 1457158. doi:10.3389/frhem.2024.1457158
CrossRef Full Text | Google Scholar
7- Ware, R. E., de Montalembert, M., Tshilolo, L., and Abboud, M. R. (2017). Sickle cell disease. Lancet 390 (10091), 311–323. doi:10.1016/S0140-6736(17)30193-9
PubMed Abstract | CrossRef Full Text | Google Scholar
8- Jastaniah, W. (2011). Epidemiology of sickle cell disease in Saudi Arabia. Ann. Saudi Med. 31 (3), 289–293. doi:10.4103/0256-4947.81540
PubMed Abstract | CrossRef Full Text | Google Scholar
9- Bin Zuair, A., Aldossari, S., Alhumaidi, R., Alrabiah, M., and Alshabanat, A. (2023). The burden of sickle cell disease in Saudi Arabia: a single-institution large retrospective study. Int. J. Gen. Med. 16, 161–171. doi:10.2147/IJGM.S393233
PubMed Abstract | CrossRef Full Text | Google Scholar
10- el-Hazmi, M. A., al-Swailem, A. R., Warsy, A. S., al-Swailem, A. M., Sulaimani, R., and al-Meshari, A. A. (1995). Consanguinity among the Saudi Arabian population. J. Med. Genet. 32 (8), 623–626. doi:10.1136/jmg.32.8.623
PubMed Abstract | CrossRef Full Text | Google Scholar
11- Kavanagh, P. L., Fasipe, T. A., and Wun, T. (2022). Sickle cell disease: a review. JAMA 328 (1), 57–68. doi:10.1001/jama.2022.10233
PubMed Abstract | CrossRef Full Text | Google Scholar
12- Salinas, C. G., and Thein, S. L. (2020). Recent advances in the treatment of sickle cell disease. Front. Physiol. 11, 435. doi:10.3389/fphys.2020.00435
PubMed Abstract | CrossRef Full Text | Google Scholar
13- pfizer. (2024). Pfizer voluntarily withdraws all lots of sickle cell disease treatment OXBRYTA® (voxelotor) from worldwide markets. Available online at: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-voluntarily-withdraws-all-lots-sickle-cell-disease (Accessed September 27, 2024).
Google Scholar
14- European Medicines Agency. (2023). Revocation of authorisation for sickle cell disease medicine adakveo. Amsterdam, The Netherlands: European Medicines Agency. Available online at: https://www.ema.europa.eu/en/news/revocation-authorisation-sickle-cell-disease-medicine-adakveo (Accessed August 30, 2024).
Google Scholar
15- Leonard, A., and Tisdale, J. F. (2024). A new frontier: FDA approvals for gene therapy in sickle cell disease. Mol. Ther. 32, 264–267. doi:10.1016/j.ymthe.2024.01.015
PubMed Abstract | CrossRef Full Text | Google Scholar
16- Rueda, J., de Miguel Beriain, Í., and Montoliu, L. (2024). Affordable pricing of CRISPR treatments is a pressing ethical imperative. CRISPR J. 7 (5), 220–226. doi:10.1089/crispr.2024.0042
PubMed Abstract | CrossRef Full Text | Google Scholar
17- Burt, T., and Dhillon, S. (2013). Pharmacogenomics in early-phase clinical development. Pharmacogenomics 14 (9), 1085–1097. doi:10.2217/pgs.13.81
PubMed Abstract | CrossRef Full Text | Google Scholar
18- Alghubayshi, A., Wijesinghe, D., Alwadaani, D., Algahtani, F. H., Abohelaika, S., Alzahrani, M., et al. (2025). Unraveling the complex genomic interplay of sickle cell disease among the Saudi population: a case-control GWAS analysis. Int. J. Mol. Sci. 26 (6), 2817. doi:10.3390/ijms26062817
PubMed Abstract | CrossRef Full Text | Google Scholar
19- Alshabeeb, M. A., Alwadaani, D., Al Qahtani, F. H., Abohelaika, S., Alzahrani, M., Al Zayed, A., et al. (2023). Impact of genetic variations on thromboembolic risk in Saudis with sickle cell disease. Genes (Basel) 14 (10), 1919. doi:10.3390/genes14101919
PubMed Abstract | CrossRef Full Text | Google Scholar
20- Tragante, V., Hemerich, D., Alshabeeb, M., Brænne, I., Lempiäinen, H., Patel, R. S., et al. (2018). Druggability of coronary artery disease risk loci. Circ. Genom Precis. Med. 11 (8), e001977. doi:10.1161/CIRCGEN.117.001977
PubMed Abstract | CrossRef Full Text | Google Scholar
21- Pushpakom, S. (2024). Introduction and historical overview of drug repurposing opportunities. Available online at: https://books.rsc.org/books/edited-volume/1885/chapter/2472263/Introduction-and-Historical-Overview-of-Drug (Accessed September 4, 2024).
Google Scholar
22- Kirkham, J. K., Estepp, J. H., Weiss, M. J., and Rashkin, S. R. (2023). Genetic variation and sickle cell disease severity: a systematic review and meta-analysis. JAMA Netw. Open 6 (10), e2337484. doi:10.1001/jamanetworkopen.2023.37484
PubMed Abstract | CrossRef Full Text | Google Scholar
23- Pincez, T., Ashley-Koch, A. E., Lettre, G., and Telen, M. J. (2022). Genetic modifiers of sickle cell disease. Hematol. Oncol. Clin. North Am. 36 (6), 1097–1124. doi:10.1016/j.hoc.2022.06.006
PubMed Abstract | CrossRef Full Text | Google Scholar
24- Page, G. P., Kanias, T., Guo, Y. J., Lanteri, M. C., Zhang, X., Mast, A. E., et al. (2021). Multiple-ancestry genome-wide association study identifies 27 loci associated with measures of hemolysis following blood storage. J. Clin. Invest 131 (13), 146077. doi:10.1172/JCI146077
PubMed Abstract | CrossRef Full Text | Google Scholar
25- Pritchard, J. L. E., O’Mara, T. A., and Glubb, D. M. (2017). Enhancing the promise of drug repositioning through genetics. Front. Pharmacol. 8, 896. doi:10.3389/fphar.2017.00896
PubMed Abstract | CrossRef Full Text | Google Scholar
26- Metaferia, B., Cellmer, T., Dunkelberger, E. B., Li, Q., Henry, E. R., Hofrichter, J., et al. (2022). Phenotypic screening of the ReFRAME drug repurposing library to discover new drugs for treating sickle cell disease. Proc. Natl. Acad. Sci. U. S. A. 119 (40), e2210779119. doi:10.1073/pnas.2210779119
PubMed Abstract | CrossRef Full Text | Google Scholar
27- Agu, P. C., Afiukwa, C. A., Orji, O. U., Ezeh, E. M., Ofoke, I. H., Ogbu, C. O., et al. (2023). Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Sci. Rep. 13 (1), 13398. doi:10.1038/s41598-023-40160-2
PubMed Abstract | CrossRef Full Text | Google Scholar
28- Telen, M. J., Malik, P., and Vercellotti, G. M. (2019). Therapeutic strategies for sickle cell disease: towards a multi-agent approach. Nat. Rev. Drug Discov.18 (2), 139–158. doi:10.1038/s41573-018-0003-2
PubMed Abstract | CrossRef Full Text | Google Scholar
29- Driss, A., Asare, K. O., Hibbert, J. M., Gee, B. E., Adamkiewicz, T. V., and Stiles, J. K. (2009). Sickle cell disease in the post genomic era: a monogenic disease with a polygenic phenotype. Genomics Insights 2009 (2), GEI.S2626–48. doi:10.4137/gei.s2626
CrossRef Full Text | Google Scholar
30- Tragante, V., Hemerich, D., Alshabeeb, M., Brænne, I., Lempiäinen, H., Patel, R. S., et al. (2018). Druggability of coronary artery disease risk loci. Circ. Genom Precis. Med. 11 (8), e001977. doi:10.1161/CIRCGEN.117.001977
PubMed Abstract | CrossRef Full Text | Google Scholar
31- Cannon, M., Stevenson, J., Stahl, K., Basu, R., Coffman, A., Kiwala, S., et al. (2024). DGIdb 5.0: rebuilding the drug-gene interaction database for precision medicine and drug discovery platforms. Nucleic Acids Res. 52 (D1), D1227–D1235. doi:10.1093/nar/gkad1040
PubMed Abstract | CrossRef Full Text | Google Scholar
32- WHO. (2024). VigiAccess. Available online at: https://www.vigiaccess.org/(Accessed September 4, 2024). Google Scholar
33- http://sideeffects.embl.de
34- Kuhn, M., Letunic, I., Jensen, L. J., and Bork, P. (2016). The SIDER database of drugs and side effects. Nucleic Acids Res. 44 (D1), D1075–D1079. doi:10.1093/nar/gkv1075
PubMed Abstract | CrossRef Full Text | Google Scholar
35- Van Bever, E., Wirtz, V. J., Azermai, M., De Loof, G., Christiaens, T., Nicolas, L., et al. (2014). Operational rules for the implementation of INN prescribing. Int. J. Med. Inf. 83 (1), 47–56. doi:10.1016/j.ijmedinf.2013.09.004
PubMed Abstract | CrossRef Full Text | Google Scholar
36- Kang, H., Pan, S., Lin, S., Wang, Y. Y., Yuan, N., and Jia, P. (2024). PharmGWAS: a GWAS-based knowledgebase for drug repurposing. Nucleic Acids Res. 52 (D1), D972–D979. doi:10.1093/nar/gkad832
PubMed Abstract | CrossRef Full Text | Google Scholar
37- Ochoa, D., Karim, M., Ghoussaini, M., Hulcoop, D. G., McDonagh, E. M., and Dunham, I. (2022). Human genetics evidence supports two-thirds of the 2021 FDA-Approved drugs. Nat. Rev. Drug Discov. 21 (8), 551. doi:10.1038/d41573-022-00120-3
PubMed Abstract | CrossRef Full Text | Google Scholar
38- Nelson, M. R., Tipney, H., Painter, J. L., Shen, J., Nicoletti, P., Shen, Y., et al. (2015). The support of human genetic evidence for approved drug indications. Nat. Genet. 47 (8), 856–860. doi:10.1038/ng.3314
PubMed Abstract | CrossRef Full Text | Google Scholar
39- Ochoa, D., Karim, M., Ghoussaini, M., Hulcoop, D. G., McDonagh, E. M., and Dunham, I. (2022). Human genetics evidence supports two-thirds of the 2021 FDA-Approved drugs. Nat. Rev. Drug Discov. 21 (8), 551. doi:10.1038/d41573-022-00120-3
PubMed Abstract | CrossRef Full Text | Google Scholar
40- Ullah, A. (2025). Perspective chapter: technological advances in population genetics. London, United Kingdom: IntechOpen. doi:10.5772/intechopen.1009271
CrossRef Full Text | Google Scholar
41- Ku, C. S., Loy, E. Y., Pawitan, Y., and Chia, K. S. (2010). The pursuit of genome-wide association studies: where are we now? J. Hum. Genet. 55 (4), 195–206. doi:10.1038/jhg.2010.19
PubMed Abstract | CrossRef Full Text | Google Scholar
42- Reay, W. R., and Cairns, M. J. (2021). Advancing the use of genome-wide association studies for drug repurposing. Nat. Rev. Genet. 22 (10), 658–671. doi:10.1038/s41576-021-00387-z
PubMed Abstract | CrossRef Full Text | Google Scholar
43- Drug Repurposing (DR) (2024). An emerging approach in drug discovery | IntechOpen. Available online at: https://www.intechopen.com/chapters/72744 (Accessed September 4, 2024).
Google Scholar
44- Nanda, H., Ponnusamy, N., Odumpatta, R., Jeyakanthan, J., and Mohanapriya, A. (2020). Exploring genetic targets of psoriasis using genome wide association studies (GWAS) for drug repurposing. 3 Biotech. 10 (2), 43. doi:10.1007/s13205-019-2038-4
PubMed Abstract | CrossRef Full Text | Google Scholar
45- Xu, J., Mao, C., Hou, Y., Luo, Y., Binder, J. L., Zhou, Y., et al. (2022). Interpretable deep learning translation of GWAS and multi-omics findings to identify pathobiology and drug repurposing in Alzheimer’s disease. Cell Rep.41 (9), 111717. doi:10.1016/j.celrep.2022.111717
PubMed Abstract | CrossRef Full Text | Google Scholar
46- Blood. (2024). ASCENT1: a phase 2 trial to evaluate the efficacy and safety of oral decitabine-tetrahydrouridine (NDec) in patients with sickle cell disease. American Society of Hematology. Available online at: https://ashpublications.org/blood/article/140/Supplement%201/5420/487531/ASCENT1-A-Phase-2-Trial-to-Evaluate-the-Efficacy(Accessed September 2, 2025).
Google Scholar
47- Chowdhury, F. A., Colussi, N., Sharma, M., Wood, K. C., Xu, J. Z., Freeman, B. A., et al. (2023). Fatty acid nitroalkenes – multi-target agents for the treatment of sickle cell disease. Redox Biol. 68, 102941. doi:10.1016/j.redox.2023.102941
PubMed Abstract | CrossRef Full Text | Google Scholar
48- Adam, S. S., and Hoppe, C. (2013). Potential role for statins in sickle cell disease. Pediatr. Blood Cancer 60 (4), 550–557. doi:10.1002/pbc.24443
PubMed Abstract | CrossRef Full Text | Google Scholar
49- Bereal-Williams, C., Machado, R. F., McGowan, V., Chi, A., Hunter, C. J., and Kato, G. J. (2012). Atorvastatin reduces serum cholesterol and triglycerides with limited improvement in vascular function in adults with sickle cell anemia. Haematologica 97 (11), 1768–1770. doi:10.3324/haematol.2011.054957
PubMed Abstract | CrossRef Full Text | Google Scholar
50- Ridker, P. M., Danielson, E., Fonseca, F. A. H., Genest, J., Gotto, A. M., Kastelein, J. J., et al. (2008). Rosuvastatin to prevent vascular events in men and women with elevated C-Reactive protein. N. Engl. J. Med. 359 (21), 2195–2207. doi:10.1056/NEJMoa0807646
PubMed Abstract | CrossRef Full Text | Google Scholar
51- Wong, K., Lai, W. K., and Jackson, D. E. (2022). HLA classII regulation of immune response in sickle cell disease patients: susceptibility to red blood cell alloimmunization (systematic review and meta-analysis). Vox Sang. 117 (11), 1251–1261. doi:10.1111/vox.13351
PubMed Abstract | CrossRef Full Text | Google Scholar
52- Tamouza, R., Neonato, M. G., Busson, M., Marzais, F., Girot, R., Labie, D., et al. (2002). Infectious complications in sickle cell disease are influenced by HLA class II alleles. Hum. Immunol. 63 (3), 194–199. doi:10.1016/s0198-8859(01)00378-0
PubMed Abstract | CrossRef Full Text | Google Scholar
53- Martins, J. O., Pagani, F., Dezan, M. R., Oliveira, V. B., Conrado, M., Ziza, K. C., et al. (2022). Impact of HLA-G +3142C>G on the development of antibodies to blood group systems other than the Rh and Kell among sensitized patients with sickle cell disease. Transfus. Apher. Sci. 61 (5), 103447. doi:10.1016/j.transci.2022.103447
PubMed Abstract | CrossRef Full Text | Google Scholar
54- Akaba, K., Nwogoh, B., Oshatuyi, O., and Angchaisuksiri, P. (2020). Determination of von Willebrand factor level in patient with sickle cell diseasein vaso-occlusive crisis. Res. Pract. Thromb. Haemost. 4 (5), 902–905. doi:10.1002/rth2.12378
PubMed Abstract | CrossRef Full Text | Google Scholar
55- Sahebkar, A., Serban, C., Ursoniu, S., Mikhailidis, D. P., Undas, A., Lip, G. Y. H., et al. (2016). The impact of statin therapy on plasma levels of von Willebrand factor antigen. Systematic review and meta-analysis of randomised placebo-controlled trials. Thromb. Haemost. 115 (3), 520–532. doi:10.1160/TH15-08-0620
PubMed Abstract | CrossRef Full Text | Google Scholar
56- Hoppe, C., Jacob, E., Styles, L., Kuypers, F., Larkin, S., and Vichinsky, E. (2017). Simvastatin reduces vaso-occlusive pain in sickle cell anaemia: a pilot efficacy trial. Br. J. Haematol. 177 (4), 620–629. doi:10.1111/bjh.14580
PubMed Abstract | CrossRef Full Text | Google Scholar
57- Xi, C., Palani, C., Takezaki, M., Shi, H., Horuzsko, A., Pace, B. S., et al. (2024). Simvastatin-mediated Nrf2 activation induces fetal hemoglobin and antioxidant enzyme expression to ameliorate the phenotype of sickle cell disease. Antioxidants (Basel) 13 (3), 337. doi:10.3390/antiox13030337
PubMed Abstract | CrossRef Full Text | Google Scholar
58- Ademi, B., Folker, J., and Rothwell, W. B. (2025). Statin-induced debilitating weakness and myopathy. Drug Ther. Bull. 63 (6), 94–97. doi:10.1136/dtb.2024.256956rep
PubMed Abstract | CrossRef Full Text | Google Scholar
59- Choi, S. A., Kim, J. S., Park, Y. A., Lee, D. H., Park, M., Yee, J., et al. (2025). Transporter genes and statin-induced hepatotoxicity. Cardiovasc Drugs Ther. 39 (4), 801–810. doi:10.1007/s10557-024-07580-2
PubMed Abstract | CrossRef Full Text | Google Scholar
60- López-López, S., Romero de Ávila, M. J., Hernández de León, N. C., Ruiz-Marcos, F., Baladrón, V., Nueda, M. L., et al. (2021). NOTCH4 exhibits anti-inflammatory activity in activated macrophages by interfering with Interferon-γ and TLR4 signaling. Front. Immunol. 12, 734966. doi:10.3389/fimmu.2021.734966
PubMed Abstract | CrossRef Full Text | Google Scholar
61- Zhou, B., Lin, W., Long, Y., Yang, Y., Zhang, H., Wu, K., et al. (2022). Notch signaling pathway: architecture, disease, and therapeutics. Signal Transduct. Target Ther. 7 (1), 95. doi:10.1038/s41392-022-00934-y
PubMed Abstract | CrossRef Full Text | Google Scholar
62- Fingerlin, T. E., Murphy, E., Zhang, W., Peljto, A. L., Brown, K. K., Steele, M. P., et al. (2013). Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 45 (6), 613–620. doi:10.1038/ng.2609
PubMed Abstract | CrossRef Full Text | Google Scholar
63- Field, J. J., Burdick, M. D., DeBaun, M. R., Strieter, B. A., Liu, L., Mehrad, B., et al. (2012). The role of fibrocytes in sickle cell lung disease. PLoS One 7 (3), e33702. doi:10.1371/journal.pone.0033702
PubMed Abstract | CrossRef Full Text | Google Scholar
64- Adeyinka, A., and Bashir, K. (2024). “Tumor lysis syndrome,” in StatPearls(Treasure Island, FL: StatPearls Publishing). Available online at: http://www.ncbi.nlm.nih.gov/books/NBK518985/
65- Pritchard, K. A. Jr, Besch, T. L., Wang, J., Xu, H., Jones, D. W., Foster, T. D., et al. (2005). Differential effects of lovastatin and allopurinol therapy in a murine model of sickle cell disease. Blood 106 (11), 2325. doi:10.1182/blood.V106.11.2325.2325
CrossRef Full Text | Google Scholar
66- Kelkar, A., Kuo, A., and Frishman, W. H. (2011). Allopurinol as a cardiovascular drug. Cardiol. Rev. 19 (6), 265–271. doi:10.1097/CRD.0b013e318229a908
PubMed Abstract | CrossRef Full Text | Google Scholar
67- Wood, K. C., Hebbel, R. P., and Granger, D. N. (2005). Endothelial cell NADPH oxidase mediates the cerebral microvascular dysfunction in sickle cell transgenic mice. FASEB J. 19 (8), 989–991. doi:10.1096/fj.04-3218fje
PubMed Abstract | CrossRef Full Text | Google Scholar
68- Kaul, D. K., Liu, X. D., Choong, S., Belcher, J. D., Vercellotti, G. M., and Hebbel, R. P. (2004). Anti-inflammatory therapy ameliorates leukocyte adhesion and microvascular flow abnormalities in transgenic sickle mice. Am. J. Physiol. Heart Circ. Physiol. 287 (1), H293–H301. doi:10.1152/ajpheart.01150.2003
PubMed Abstract | CrossRef Full Text | Google Scholar
69- Dashti, M., Al-Matrouk, A., Channanath, A., Hebbar, P., Al-Mulla, F., and Thanaraj, T. A. (2022). Distribution of HLA-B alleles and haplotypes in Qatari: recommendation for establishing pharmacogenomic markers screening for drug hypersensitivity. Front. Pharmacol. 13, 891838. doi:10.3389/fphar.2022.891838
PubMed Abstract | CrossRef Full Text | Google Scholar
70- Dean, L., and Kane, M. (2012). “Allopurinol therapy and HLA-B*58:01 genotype,” in Medical genetics summaries. Editors V. M. Pratt, S. A. Scott, M. Pirmohamed, B. Esquivel, B. L. Kattman, and A. J. Malheiro (Bethesda, MA: National Center for Biotechnology Information US). Available online at: http://www.ncbi.nlm.nih.gov/books/NBK127547/
71- Yessayan, L. T., Neyra, J. A., Westover, A. J., Szamosfalvi, B., and Humes, H. D. (2022). Extracorporeal immunomodulation treatment and clinical outcomes in ICU COVID-19 patients. Crit. Care Explor 4 (5), e0694. doi:10.1097/CCE.0000000000000694
PubMed Abstract | CrossRef Full Text | Google Scholar
72- Rees, D. C., Kilinc, Y., Unal, S., Dampier, C., Pace, B. S., Kaya, B., et al. (2022). A randomized, placebo-controlled, double-blind trial of canakinumab in children and young adults with sickle cell anemia. Blood 139 (17), 2642–2652. doi:10.1182/blood.2021013674
PubMed Abstract | CrossRef Full Text | Google Scholar
73- Drugs.com (2024). Xolair (omalizumab) FDA Approval History. Available online at: https://www.drugs.com/history/xolair.html
74- Kaplan, A. P., Giménez-Arnau, A. M., and Saini, S. S. (2017). Mechanisms of action that contribute to efficacy of omalizumab in chronic spontaneous urticaria. Allergy 72 (4), 519–533. doi:10.1111/all.13083
PubMed Abstract | CrossRef Full Text | Google Scholar
75- Benjamin, E. J., Dupuis, J., Larson, M. G., Lunetta, K. L., Booth, S. L., Govindaraju, D. R., et al. (2007). Genome-wide association with select biomarker traits in the framingham heart study. BMC Med. Genet. 8 (Suppl. 1), S11. doi:10.1186/1471-2350-8-S1-S11
PubMed Abstract | CrossRef Full Text | Google Scholar
76- Al-Suliman, A., Elsarraf, N. A., Baqishi, M., Homrany, H., Bousbiah, J., and Farouk, E. (2006). Patterns of mortality in adult sickle cell disease in Al-Hasa region of Saudi Arabia. Ann. Saudi Med. 26 (6), 487–488. doi:10.5144/0256-4947.2006.487
PubMed Abstract | CrossRef Full Text | Google Scholar
77- Alhaj, Z. M., Mohamed, N. E., Mady, A. F., Alamri, M. M., Alshammari, S., Alshebaily, A. K., et al. (2023). Predictors of mortality in adults with sickle cell disease admitted to the intensive care unit in king saud medical city, Saudi Arabia. Cureus 15 (5), e38817. doi:10.7759/cureus.38817
PubMed Abstract | CrossRef Full Text | Google Scholar
78- An, P., Barron-Casella, E. A., Strunk, R. C., Hamilton, R. G., Casella, J. F., and DeBaun, M. R. (2011). Elevation of IgE in children with sickle cell disease is associated with doctor diagnosis of asthma and increased morbidity. J. Allergy Clin. Immunol. 127 (6), 1440–1446. doi:10.1016/j.jaci.2010.12.1114
PubMed Abstract | CrossRef Full Text | Google Scholar
79- Awojoodu, A. O., Keegan, P. M., Lane, A. R., Zhang, Y., Lynch, K. R., Platt, M. O., et al. (2014). Acid sphingomyelinase is activated in sickle cell erythrocytes and contributes to inflammatory microparticle generation in SCD. Blood 124 (12), 1941–1950. doi:10.1182/blood-2014-01-543652
PubMed Abstract | CrossRef Full Text | Google Scholar
80- Fernández-Ruiz, M., and Aguado, J. M. (2018). Risk of infection associated with anti-TNF-α therapy. Expert Rev. Anti Infect. Ther. 16 (12), 939–956. doi:10.1080/14787210.2018.1544490
PubMed Abstract | CrossRef Full Text | Google Scholar
81- Contejean, A., Janssen, C., Orsini-Piocelle, F., Zecchini, C., Charlier, C., and Chouchana, L. (2023). Increased risk of infection reporting with anti-BCMA bispecific monoclonal antibodies in multiple myeloma: a worldwide pharmacovigilance study. Am. J. Hematol. 98 (12), E349–E353. doi:10.1002/ajh.27071
PubMed Abstract | CrossRef Full Text | Google Scholar
82- Solovey, A., Somani, A., Belcher, J. D., Milbauer, L., Vincent, L., Pawlinski, R., et al. (2017). A monocyte-TNF-endothelial activation axis in sickle transgenic mice: therapeutic benefit from TNF blockade. Am. J. Hematol. 92 (11), 1119–1130. doi:10.1002/ajh.24856
PubMed Abstract | CrossRef Full Text | Google Scholar
83- Adelowo, O., and Edunjobi, A. S. (2011). Juvenile idiopathic arthritis coexisting with sickle cell disease: two case reports. BMJ Case Rep. 2011, bcr1020114889. doi:10.1136/bcr.10.2011.4889
PubMed Abstract | CrossRef Full Text | Google Scholar
84- Drugs.com(2024a). Simvastatin Prices, Coupons & Patient Assistance Programs. Available online at: https://www.drugs.com/price-guide/simvastatin(Accessed September 4, 2024).
Google Scholar
85- Drugs.com(2024b). Allopurinol Prices, Coupons & Patient Assistance Programs. Available online at: https://www.drugs.com/price-guide/allopurinol(Accessed September 4, 2024).
Google Scholar
86- Drugs.com(2024c). Ilaris Prices, Coupons & Patient Assistance Programs. Available online at: https://www.drugs.com/price-guide/ilaris(Accessed September 4, 2024).
Google Scholar
87- Drugs.com (2024d). Xolair Prices, Coupons & Patient Assistance Programs. Available online at: https://www.drugs.com/price-guide/xolair (Accessed September 4, 2024).
Google Scholar
88- Uchil, P. D., Hinz, A., Siegel, S., Coenen-Stass, A., Pertel, T., Luban, J., et al. (2013). TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J. Virol. 87 (1), 257–272. doi:10.1128/JVI.01804-12
PubMed Abstract | CrossRef Full Text
89- Qian, D., Cong, Y., Wang, R., Chen, Q., Yan, C., and Gong, D. (2023). Structural insight into the human SID1 transmembrane family member 2 reveals its lipid hydrolytic activity. Nat. Commun. 14 (1), 3568. doi:10.1038/s41467-023-39335-2
PubMed Abstract | CrossRef Full Text | Google Scholar
90- Geng, M. Y., Wang, L., Song, Y. Y., Gu, J., Hu, X., Yuan, C., et al. (2021). Sidt2 is a key protein in the autophagy-lysosomal degradation pathway and is essential for the maintenance of kidney structure and filtration function. Cell Death Dis. 13 (1), 7. doi:10.1038/s41419-021-04453-6
PubMed Abstract | CrossRef Full Text | Google Scholar
91- Estelius, J., Lengqvist, J., Ossipova, E., Idborg, H., Le Maître, E., Andersson, M. L. A., et al. (2019). Mass spectrometry-based analysis of cerebrospinal fluid from arthritis patients—Immune-related candidate proteins affected by TNF blocking treatment. Arthritis Res. Ther. 21, 60. doi:10.1186/s13075-019-1846-6
PubMed Abstract | CrossRef Full Text | Google Scholar
92- Jarczak, J., Thetchinamoorthy, K., Wierzbicka, D., Bujko, K., Ratajczak, M. Z., and Kucia, M. (2025). Expression of innate immunity genes in human hematopoietic stem/progenitor cells – single cell RNA-Seq analysis. Front. Immunol. 16, 1515856. doi:10.3389/fimmu.2025.1515856
PubMed Abstract | CrossRef Full Text | Google Scholar
المصدر الرئيس:
https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2025.1671626/full

